Thromboxane A2 Deficit Diagnosis in a Patient with Suspicion of Von Willebrand Disease
A B S T R A C T
Thromboxane A2 receptor deficiency is a qualitative platelet disorder that partially impedes adequate platelet signaling and aggregation. Generally, these patients have mild hemorrhagic manifestations in basal conditions, but may show severe bleeding when faced with a hemostatic challenge. We present the case of a 30-year-old female patient that arrives at the Hematology service with an undiagnosed bleeding disorder. She presented hemorrhagic complications during an augmentation mammoplasty and during an exodontia procedure, yet, during a C-section she presented none. At the first consultation she had normal coagulation factor levels by coagulometry, and normal platelet aggregation test for ADP, ristocetin, collagen and epinephrine. A platelet aggregation test for arachidonic acid confirmed thromboxane A2 deficit disorder. Thromboxane A2 deficit disorder is a rare qualitative platelet disorder that requires an extensive knowledge of coagulation and platelet function and tests, and a high level of clinical suspicion. These tests are especially difficult to interpret during pregnancy due to normal modifications to bodily function during this process.
Keywords
Thromboxane A2 Receptor, thromboxane A2 receptor deficiency, thrombocytopathy, blood platelet disorder
Introduction
Qualitative thrombocytopathies are uncommon disorders that present a diagnostic challenge. They require a high level of suspicion, and common disorders must be discarded first. Pregnancy is not an ideal moment to evaluate blood dyscrasias due to its inherent prothrombotic nature, especially from the end of the second trimester onwards where partial deficits may be hidden. In certain situations, it’s difficult to establish a definitive diagnosis due to the high cost and complexity of some confirmatory tests like platelet aggregation, flow cytometry and genetic tests.
Case Presentation
We present the case of a 30-year-old female patient, with a history of mayor bleeding in an exodontia procedure and an augmentation mammoplasty. Her presumptive diagnosis to this point had been Von Willebrand disease due to decreased factor VIII levels at 45%. At the moment of the initial consultation, she was 24 weeks pregnant, asymptomatic, without any complications or bleeding. The coagulation studies during the end of the second trimester were not conclusive. The pregnancy was terminated by cesarean-section without any complications. The next consultation was two years later, to that point she hadn’t had any hemorrhagic manifestations, complete exams were solicited. The coagulometry tests and platelet aggregation test for ADP, collagen, ristocetin and epinephrine came back normal (Table 1), as well as the VIII factor and the Von Willebrand factor. Finally, an abnormal report of platelet adhesion tests with arachidonic acid showed suppression of aggregation with escalating concentrations which lead to a diagnosis of thromboxane A2 receptor deficit (Table 2).
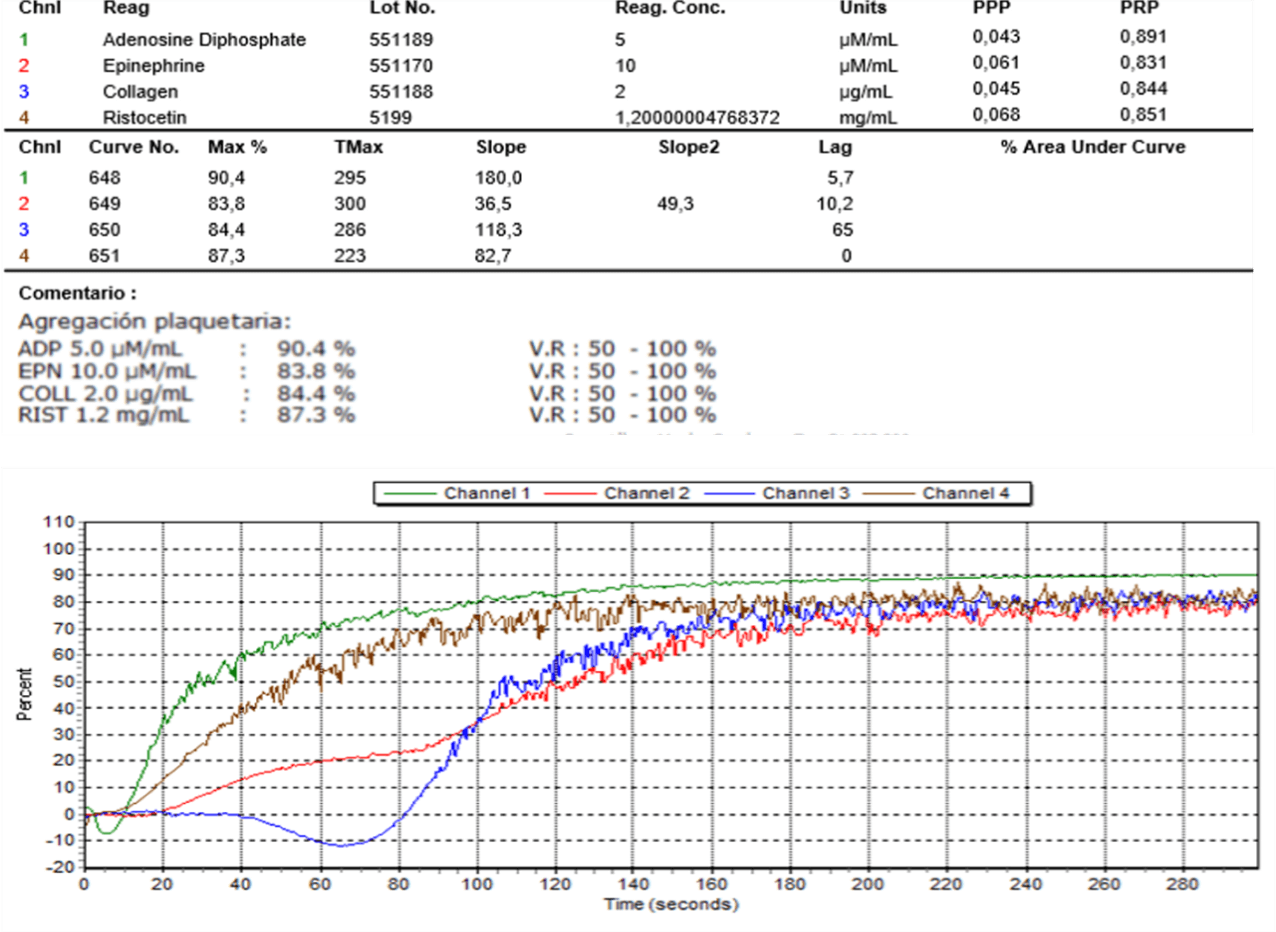
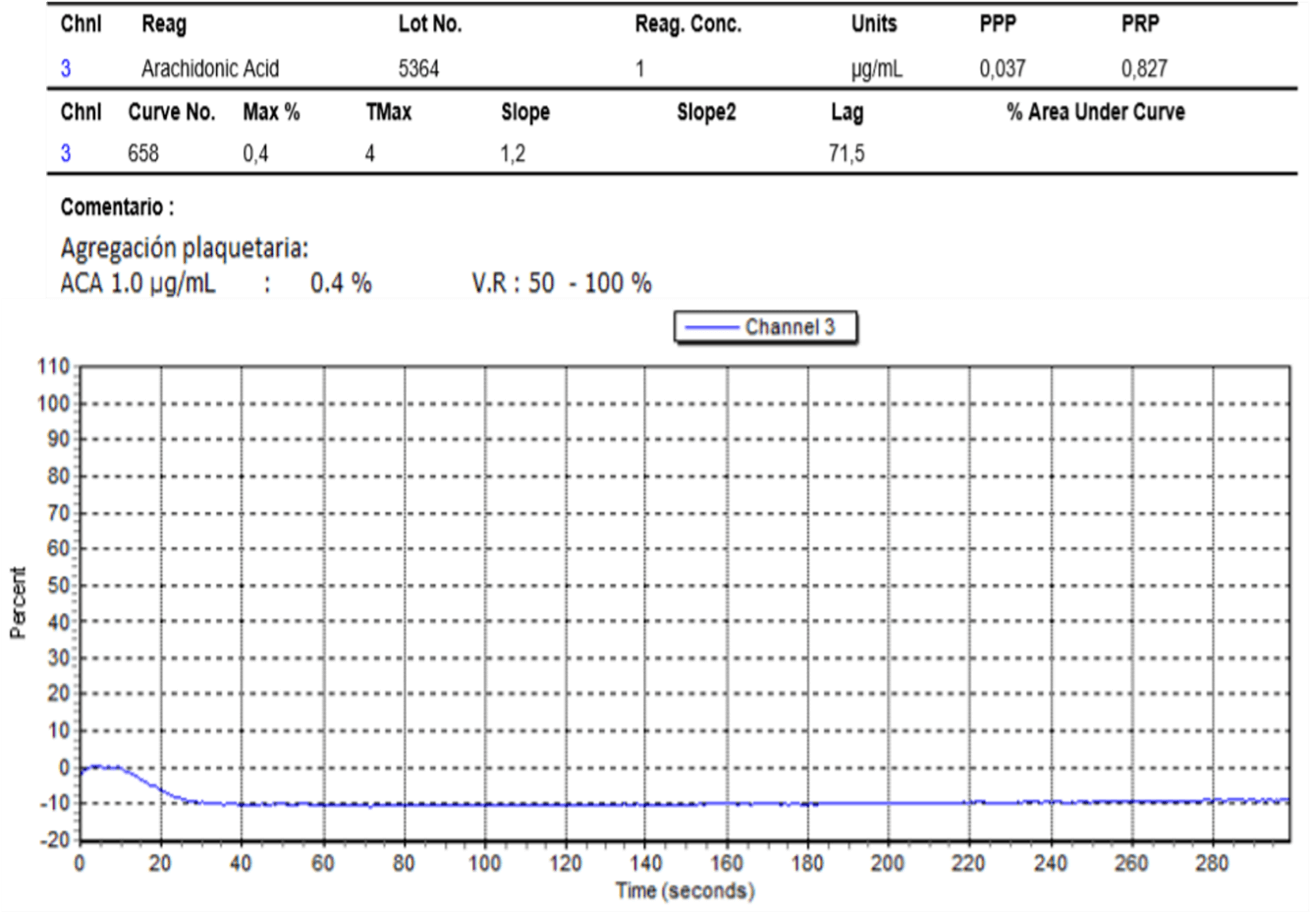
Discussion
Platelet activation is a complex act mediated by biochemical processes that lead to structural modifications to the platelet so it can aggregate and secrete substances. This process may be induced by various physiological agonists such as adenosine diphosphate (ADP), the platelet activating factor (4FAP) and thromboxane A2 (TXA2) [1]. TXA2 is an eicosanoid derived from arachidonic acid produced in the platelet through the action of cyclooxygenase enzyme 1 (COX1), it has a short half-life, with an autocrine effect over platelets achieving platelet aggregation, granule liberation and vasoconstriction [1-3].
The thromboxane A2 receptor is a key molecule in hemostasis. Two isoforms have been described: placental and endothelial TXA2 receptor. These receptors are part of the protein-G-coupled receptors, specifically Gq, related to phospholipase C, diacylglycerol and inositol 1,4,5-triphosphate [1]. When the TXA2 receptor disfunctions it becomes uncapable of forming adequate dimers with the protein-G-coupled receptor making the signaling process impossible [4]. The first mutations described in TXA2 receptor was a change of arginine for leucine in the 60th location in the first cytoplasmic loop, which is a highly conserved region in the protein-G-coupled receptors [1, 5, 6]. This generates a platelet–agonist interaction defect. TXA2 production is normal in this context [7]. To this day six mutations of the TXA2 receptor are known, five qualitative and one quantitative [3, 8]. Both autosomic dominant and recessive presentations have been described sometimes affecting aggregation through TXA2 and other agonists concomitantly, such as collagen and thrombin. Heterozygous patients will generally present low platelet aggregation levels [6].
The clinical context of a TXA2 receptor deficit is similar to the administration of acetylsalicylic acid (ASA). ASA will irreversibly link to the COX-1, acetylating a serine residue reducing the production of TXA2, leading to a reduced stimulation of the platelet’s TXA2 receptor [2, 3, 8]. The TXA2 receptor deficit diagnosis requires an abnormal platelet aggregation test with arachidonic acid, generally performed after platelet aggregation tests with ADP, collagen and ristocetin are reported as normal in three opportunities. Clinical manifestations of platelet partial signalization defects are generally mild in basal conditions, but faced with a hemostatic challenge, such as the case of surgery, will present severe bleeding [9]. Additionally, pregnancy presents an additional challenge: Some procoagulant factors will increase during pregnancy such as Von Willebrand factor, fibrinogen, factor VII, VIII and X; factors II, V, IX and XII remain stable; factor XIII decreases. Additionally, fibrinolytic activity is decreased during the preparation for the hemostatic challenge of labor with an increase in the plasminogen activation factor inhibitor I and II, and a decrease in free protein S [10].
This context that favors timely hemostasis in the postpartum may explain why this patient didn’t present hemorrhagic complications in this period. There is little information of patients with TXA2 deficit during pregnancy, yet, hemorrhagic complications during pregnancy have been described [11]. On the other hand, the initial laboratories with a discrete lowering of the von Willebrand factor led to the suspicion of Von Willebrand disease. This factor is a central component of hemostasis, that serves in the adhesion between platelets and with injured vascular endothelium and as a transporter of factor VIII. Anomalies of the von Willebrand factor may lead to von Willebrand disease, the most common hereditary hemorrhagic disorder. It occurs in 1 of 1000 patients and it is associated with a quantitative deficiency (Type 1 and 3) or a qualitative anomaly (Type 2) [12]. This possibility was discarded when the von Willebrand and factor VIII levels were normal posteriorly.
Another differential diagnosis to consider is Scott syndrome, an uncommon hemorrhagic syndrome where the phosphatidyl-serine molecule is less active leading to a diminished expression of this molecule on the platelet surface. There is a diminished capacity to form tenase, the prothrombinase complex and the formation of thrombin leading to hemorrhagic complications. Generally, the abnormal platelet aggregation tests make this diagnosis less probable, but to confirm the diagnosis flow cytometry with a decreased thrombin stimulation is required [13]. We do not possess this kind of technology at the moment at our center, yet, the Scott syndrome if accompanied with other phenotypic abnormalities that the patient did not have.
Conclusion
The diagnosis of qualitative thrombocytopathies is still a challenge, it requires a high level of clinical suspicion. Additionally, pregnancy is a procoagulant state, thus coagulation tests done in this context must be interpreted carefully. One must know which test to order, how to interpret them and have the resources to do so. In patients with severe bleeding when challenged, with a normal hemogram, normal coagulation and coagulometric tests it is important to think about qualitative thrombocytopathies as a serious diagnostic possibility.
Consent
Written informed consent was obtained from the patient for publication of this case report and accompanying tables.
Conflicts of Interest
None.
Article Info
Article Type
Case ReportPublication history
Received: Fri 29, Jan 2021Accepted: Tue 09, Feb 2021
Published: Fri 26, Feb 2021
Copyright
© 2023 Francisco Jaramillo. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited. Hosting by Science Repository.DOI: 10.31487/j.TCR.2021.01.01
Figures & Tables
References
1. Hirata T, Ushikubi F, Kakizuka A, Okutna M, Narumiya S (1996) Two thromboxane A2 receptor isoforms in human platelets: Opposite coupling to adenylyl cyclase with different sensitivity to Arg60 to Leu mutation. J Clin Invest 97: 949-956. [Crossref]
2.
Cantley L (2012)
Signal Transduction. Medical Physiology A Cellular and Molecular Approach.
Second Edition. Philadelphia. Elsevier 48-74.
3.
Mundell SJ,
Mumford A (2018) TBXA2R gene variants associated with bleeding. Platelets 29:
739-742. [Crossref]
4.
Capra V, Mauri M,
Guzzi F, Busnelli M, Accomazzo MR et al. (2017) Impaired thromboxane receptor
dimerization reduces signaling efficiency: A potential mechanism for reduced
platelet function in vivo. Biochem Pharmacol 124: 43-56. [Crossref]
5.
Hirata T,
Kakizuka A, Ushikubi F, Fuse I, Okuma M et al. (1994) Arg60 to Leu mutation of
the human thromboxane A2 receptor in a dominantly inherited bleeding disorder. J
Clin Invest 94: 1662-1667. [Crossref]
6.
Rao AK (2003)
Inherited defects in platelet signaling mechanisms. J Thromb Hemost 1:
671-681. [Crossref]
7.
Rao AK, Coller BS
(2016) Hereditary Qualitative Platelet Disorders. Williams’s Hematology. 9th
Edition. New York: Mc Graw Hill 2039-2072.
8.
Bugert P, Fischer
L, Althaus K, Knöfler R, Bakchoul T (2020) Platelet dysfunction caused by a
novel thromboxane A2 receptor mutation and congenital thrombocytopenia in a
case of mild bleeding. Platelets 31: 276-279. [Crossref]
9.
Rao AK, Gabbeta J
(2000) Congenital disorders of platelet signal transduction. Arterioscler
Thromb Vasc Biol 20: 285-289. [Crossref]
10. Mims MP (2016) Hematology During Pregnancy. Williams’s
Hematology. 9th Edition. New York: Mc Graw Hill 119-128.
11. Civaschi E, Klersy C, Melazzini F, Pujol Moix N,
Santoro C et al. (2015) Analysis of 65 pregnancies in 34 women with five
different forms of inherited platelet function disorders. Br J Haematol
170: 559-563. [Crossref]
12. Johnsen J, Ginsburg D (2016) Von Willebrand Disease.
Williams’s Hematology. 9th edition. New York: Mc Graw Hill 2166-2176.
13. Flores Nascimento MC, Orsi FLA, Yokoyama AP, Pereira
FG, Lorand Metze I et al. (2012) Diagnosis of Scott syndrome in patient with
bleeding disorder of unknown cause. Blood Coagul Fibrinolysis 23: 75-77.
[Crossref]