The Pivotal Role of PAO in CDK Inhibitors (Roscovitine and Purvalanol)- Triggered Apoptosis in PUMA Null HCT116 Colon Cancer Cells
A B S T R A C T
Background: Cycline-dependent kinase inhibitors (CDKi); roscovitine and purvalanol, are promising anti-cancer drugs due to their strong anti-proliferative effectiveness due to activation of PA catabolism. Besides transforming acetylated spermine and spermidine into spermidine and putrescine, respectively, polyamine oxidase (PAO) also generates hydrogen peroxide in high concentrations as a by-product. PAO was assumed as a pivotal key molecule during drug-induced apoptosis in cancer cells. Our aim is to reveal the role of PAO action in CDKi-triggered apoptosis in Puma knock-out HCT116 colon cancer cells.
Methods: HCT116 wt and HCT116 Puma-/- cells were treated with Roscovitine and Purvalanol and cell viability and apoptosis were determined. Protein was isolated from treated and untreated cells and key molecules of cell cycle control and polyamine pathways were investigated at translational level. Polyamine content was determined by HPLC for all conditions. MDL-72527 was used as a PAO inhibitor and apoptotic cell death was analysed.
Results: Roscovitine and purvalanol induced apoptosis and increased the cytotoxic responses in HCT116 wt and HCT116 Puma-/- colon carcinoma cell lines by modulating CDK1, 4, cyclin-B1, D3. Both, CDKi altered intrinsic apoptotic pathways in HCT116 wt. Whereas, drug-induced apoptosis occurred caspase-independent in Puma-/- colon cancer cells. Roscovitine and purvalanol up-regulated polyamine catabolic enzymes, whereas CDK inhibitors decreased the polyamine levels in HCT116 wt and HCT116 Puma-/- colon cancer cells. In addition, PAO inhibitor MDL72527 prevented drug-induced apoptosis.
Conclusion: PAO expression profile might be a critical target in CDK inhibitors-triggered apoptosis in HCT116 colorectal cancer cells. Thus, MAPK signaling pathway relations with cell cycle and polyamine catabolic pathway investigations are in progress.
Keywords
Puma, colon cancer, CDK inhibitors, polyamine, apoptosis
Introduction
Colorectal cancer is an important component of the prevalence and mortality of cancer worldwide. It is the fourth common frequent type of cancer and ranked at third place in cancer-related deaths globally [1]. Conventionally, chemotherapeutic agents such as oxaliplatin and 5-FU are used for the treatment of metastatic type, but targeted therapies which particularly inhibit cell proliferation are drawing attention for chemotherapy [2]. Cyclin-dependent kinases (CDKs) regulate the cell cycle machinery in eukaryotic cells in that, they bind to specific targets, called cyclins, and lead to phosphorylation of key proteins essentially involved in DNA synthesis. CDKs make complexes with cyclins and these complexes regulate important processes like growth, proliferation and differentiation [3]. Novel CDK inhibitors purvalanol and roscovitine, have been shown to prevent CDKs from binding to cyclins and thus induce apoptosis via arrest of the cell cycle in multiple myeloma, prostate, lung, breast and colorectal cancer [4-8]. The polycationic amine derivatives, spermine, spermidine and putrescine, which are referred as natural polyamines (PAs), regulate cellular proliferation and also differentiation thus, induce tumoral growth [9]. Since PAs are cationic, they protect the cells from apoptosis and by this way, in malign cells, PA levels were elevated [10].
Accordingly, the enzyme which plays a role in PA biosynthesis, ornithine decarboxylase (ODC), was overexpressed in cancer cells [10]. On the other hand, PA catabolism converts PAs into oxidized derivatives and causes the accumulation of toxic substances. Thereby, activated polyamine catabolism improves the effects of therapeutics. The levels of PAs in the cell are controlled by enzymes of PA catabolism, spermidine/spermine N1-acetyltransferase (SSAT), spermine oxidase (SMO) and acetyl polyamine oxidase (PAO) [11]. In this study, we aimed to demonstrate the effects of purvalanol and roscovitine on apoptotic pathways and examine their roles in polyamine catabolism in HCT116 wild type and HCT116 Puma knockout colorectal cancer cells.
Materials and Methods
I Drugs and Cell Lines
Roscovitine and Purvalanol were purchased from Sigma (St. Louis, MO, USA) and Tocris Bioscience (Bristol, United Kingdom), respectively. Both were dissolved in DMSO and 10 mM stock solution was prepared. HCT116 (CCL-247) cells were purchased from American Type Culture Collection (ATCC, Manassas, USA). HCT116 Puma−/− cells were a kind gift of Bert Vogelstein (Howard Hughes Medical Institute, Johns Hopkins University, USA). HCT116 wt and Puma-/- cells were grown in McCoy’s5A medium (PAN Biotech, Aidenbach, Germany) supplemented with 10% fetal bovine serum (PAN Biotech), 100 U/100 mg.ml-1 penicillin-streptomycin, 2 mM L-glutamine, 1% nonessential amino acids (Biological Industries, Kibbutz Beit-Haemek, Israel) in 5% CO2 present in 37°C incubator.
II Determination of Cell Viability by MTT Assay
In order to examine the effect of CDKi on cell viability, wt and Puma-/- cells were seeded in 96-well plates as 1×104 cells/well. After overnight incubation, purvalanol and roscovitine were applied to cells in a dose-dependent manner (0-50 µM for roscovitine and 0-30 µM for purvalanol). After 24 h, 10 µl MTT solution (3-(4,5dimethylthiazol-2-yl)-2,5-diphenyl-tetrazolium bromide) which was prepared in PBS as 5 mg/ml stock was added and incubated for 4 h at 37°C incubator. When incubation overs, media were aspirated and 200 µl/well DMSO was used to dissolve the formazan crystals. Absorbance at 570 nm/655 nm dual-wavelength were measured and data were analysed by GraphPad Prism 8.
III Cell Death ELISA Assay for Apoptosis Determination
Cell death Detection ELISA Plus kit (Roche, Indianapolis, USA) was used to determine cell death as manufacturer’s guidelines. In brief, 1×105 cells were seeded per well in 96-well plates. After the overnight incubation, cells were treated with purvalanol and roscovitine for 24 h. Then, the cells were lysed, and the lysates were added to the microplate coated with streptavidin. Anti-DNA POD and anti-histone-biotin mixture were also added to the wells and incubated at room temperature for two hours. After incubation, wells were washed and absorbance at 405 nm wavelength was measured.
IV Annexin V-FITC/PI Staining
HCT116 wt and Puma-/- cells were seeded on 6-well plates with 3×105 cells/well density and treated with Purvalanol (15 µM and 30 µM) and Roscovitine (20 µM and 50 µM). After 24 h, the apoptotic cells were determined by Annexin V/ PI analysis [8].
V Propidium Iodide (PI), DiOC6 and H2DCFDA Staining
1×104 cells/wells HCT116 wt and Puma-/- cells seeded at 12-well plate, following cell attachment, cells were treated with purvalanol (30 μM) and roscovitine (50 μM) for 24 h. Then cells were stained by PI (2 µg/ml) for 30 min, DiOC6 (4 nM) for 5 min and H2DCFDA (0.5 µM) for 15 min, respectively. Cells were visualized under fluorescence microscope (Olympus, Japan) with appropriate filter (Ex/Em = 493 nm/636 nm for PI staining), (Ex/Em = 488 nm/525 nm for DiOC6 staining), (Ex/Em: ~492–495/517–527 nm for H2DCFDA staining).
VI Extracting Total Protein from cells and Immunoblotting
After incubation of both wt and Puma-/- HCT116 cells with roscovitine and purvalanol for 24 h, each cellular extract was performed according to our previous study [8].
VII Determination of Polyamines by HPLC
For the determination of intracellular polyamine levels, benzoylation was carried out by High-Performance Liquid Chromatography (HPLC) [8].
VIII Statistical Analysis
All the experiments were quantified by GraphPad Prism 6 Software (CA, USA) and statistically analysed by a two-way ANOVA. Statistically significant results by ANOVA were further analysed by Bonferroni’s test (where indicated). A p-value < 0.05 was considered statistically significant. Error bars in the graphs were generated using ± standard deviation (SD) values from independent experiments. Western Blot results were repeated at least twice, and band intensities were calculated by Image J program (Link).
Results
I Cdk Inhibitors Roscovitine and Purvalanol Induce Cell Viability Loss and Cell Death in HCT116 Cells in Increasing Doses
To determine the cytotoxicity of CDK inhibitors Roscovitine and Purvalanol on HCT116 colorectal carcinoma cells, the cells were treated with increasing doses (0-75 µM) of roscovitine and purvalanol for 24 hours and the relative cell viability was determined by MTT cell viability test. According to the MTT assay, roscovitine and purvalanol treatment decreased the viability of HCT116 cells in a dose-dependent manner relative to untreated control cells. 20 µM and 50 µM Roscovitine decreased the cell viability by 75% and 51%, respectively. 15 µM Purvalanol treatment decreased the viability of the cells by 67%, while 30 µM Purvalanol treatment decreased by 49% (Figure 1A). Roscovitine-induced cell death was determined by propidium iodide staining of the cells and a dose-dependent increase in PI-stained cells was observed under fluorescent microscope after Roscovitine treatment (Figure 1B).

Figure 1: Effects of CDKi on HCT116 colon cancer cells. A) The dose-dependent effect of CDK inhibitors on cell viability was determined by MTT cell viability assay after roscovitine or purvalanol (0-75 μM) treatments for 24 h. B) HCT116 wt cells were treated with 0-100 µM roscovitine for 24 h and stained by PI dye and CDKi induced cell death was visualized by PI staining at fluorescence microscopy. UT: Untreated control cells, Magnification: 400X. C) Cytotoxicity of CDK inhibitors on HCT116 Puma-/- cells were determined by MTT assay. D) HCT116 wt and Puma-/- cells were treated with Roscovitine (50 µM) or Purvalanol (30 µM) for 24 h and examined under fluorescent microscopy after propidium iodide staining. The columns represent the mean ±S.D. Two independent trials with at least 2 replicates. Statistical difference was analysed using an unpaired t-test; *p<0.05.
On the other hand, Puma deficient cells, HCT116 Puma-/-, demonstrated more resistance than wild type cells against 50 µM Roscovitine (51% and 59% in HCT116 and HCT116 Puma-/- cells, respectively) and 30 µM Purvalanol treatment (50% and 62% in HCT116 and HCT116 Puma-/- cells, respectively) (Figure 1C) in MTT assay. The same resistance profile was observed in propidium iodide staining of the cells after 50 µM Roscovitine treatment (Figure 1D). Fluorescent images indicated induction of cellular death by roscovitine or purvalanol treatment in both wt and Puma-/- cells.
II Roscovitine and Purvalanol Treatment Induce Cell Death in Both HCT116 and HCT116 Puma-/- Knockout Cells via Caspase-Dependent Apoptosis
For specification of cell death type, we primarily performed Annexin V-FITC/PI labeling following roscovitine (20 µM and 50 µM) or purvalanol (15 µM and 30 µM) treatment. As seen in (Figure 2A), FACS flow analysis of Annexin V-FITC/PI staining indicates that 20 µM and 50 µM Roscovitine treatment increased Annexin V+/PI+ cell population by approximately 2.5 and 4-fold, respectively. Although Puma deficient cells showed resistance in late apoptotic induction with respect to wt cells, about 3-fold increase in Annexin V+ apoptotic cells were observed. On the other hand, Purvalanol treatment significantly induced PI+ cell population in HCT116 wt cells, whereas necrotic induction is much less observed in Puma deficient cells. Since procaspase-9 and subsequent PARP cleavage is a cellular marker of apoptosis, we checked cleaved PARP and cleaved caspase-9 protein levels by immunoblotting. Results indicate that Roscovitine (20 µM and 50 µM) or Purvalanol (15 µM and 30 µM) treatment triggered PARP and procaspase-9 cleavage in both colorectal carcinoma cells. As mitochondrial membrane disruption has been shown to participate in apoptotic induction, we stained both colorectal cancer cells with DiOC6 after CDKi treatment and visualized by fluorescent microscope and revealed by flow cytometry (Figure 3A). Along with the obvious decrease in stained cell population after CDK inhibitors treatment, flow cytometry analysis specified MMP loss to 36% in HCT116 wt cells and to 42% in Puma deficient cells relative to control cells (100%) by roscovitine (50 µM) treatment.
In addition, 30 µM Purvalanol indicated 48% and 49% MMP loss in wt and Puma-/- cells, respectively (Figure 3B). In order to further designate the CDKi-induced caspase-dependent apoptosis induction, caspase-3 inhibitor Z-DEVD-FMK and caspase-9 inhibitor Z-LEHD-FMK was used along with CDK inhibitors and MTT assay was performed. As shown in (Figure 3C), in HCT 116 wt cells, caspase inhibitor treatment along with Roscovitine (50 µM) recovered viable cells from 74-90% for Z-DEVD-FMK and to 104% for Z-LEHD-FMK whereas Purvalanol-caspase inhibitor co-treatment recovered viable cells from 40-59% for Z-DEVD-FMK and to 62% for Z-LEHD-FMK. In Puma-/- cells, recovery percentage is less than wt cells but yet, an increase from 51-58% for Z-DEVD-FMK and to 64% for Z-LEHD-FMK in co-treatment with Roscovitine was observed (Figure 3D).
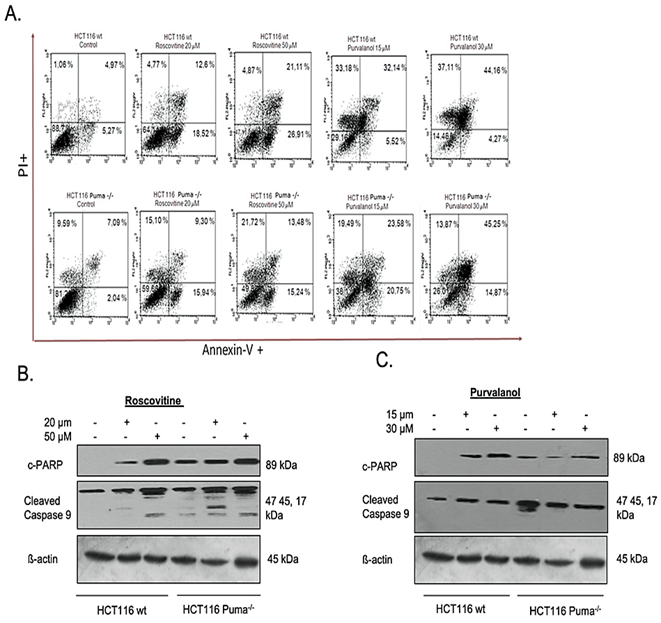
Figure 2: Determination of apoptosis induction by CDK inhibitors in HCT116 wt and Puma-/- cells. A) Apoptotic cell populations were determined by Annexin V-FITC/PI staining. HCT116 wt and Puma-/- cells were seeded in a six-well plate, treated with Roscovitine (20 µM and 50 µM) or Purvalanol (15 µM and 30 µM). Cells were then stained with Annexin V-FITC and PI and fluorescence signals were detected by flow cytometry. X-axis and Y-axis represent the Annexin V-FITC and PI signals, respectively. PARP cleavage and caspase-9 activation were determined by immunoblotting in B) Roscovitine (20 or 50 µM) and C) Purvalanol (15 or 30 µM) treated HCT116 wt and Puma-/- colorectal carcinoma cells. ß-actin was used as a loading control.
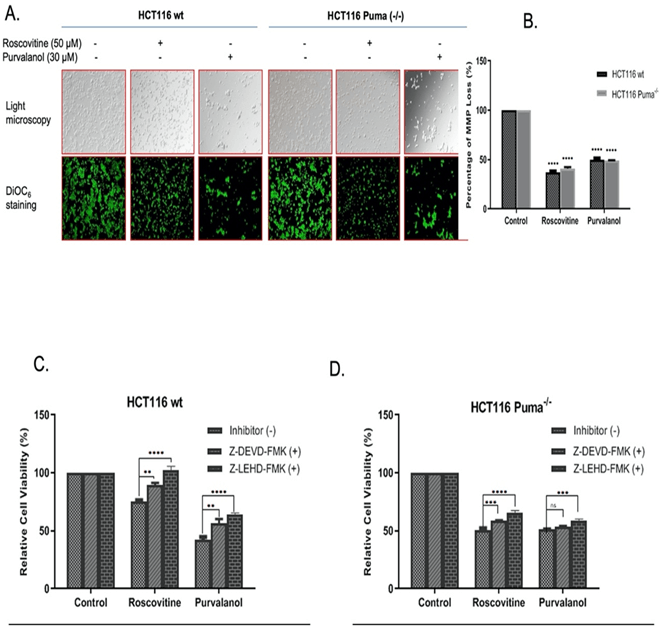
Figure 3: CDKi induced MMP loss in both colorectal carcinoma cell lines. A) The disruption of Δψm was visualized by fluorescence microscopy (200x) and B) Δψm loss were measured via flow cytometry. (Ex.= 485nm Em. =538 nm) after DiOC6 staining. Caspase-dependent CDKi-induced apoptotic cell death was demonstrated by MTT test via using caspase-3 and -9 inhibitors Z-DEVD-FMK and Z-LEHD-FMK, respectively. Following 1 h pre-treatment of cells with Z-DEVD-FMK (10 µM) and Z-LEHD-FMK (10 µM), C) HCT116 wt, D) HCT116 Puma-/- cells were treated with each CDK inhibitors for 24 h. Increase in the cell viability following Z-DEVD-FMK and Z-LEHD-FMK co-treatment were determined by MTT cell viability assay. Fluorometer results were obtained from two different culture conditions with at least 4 replicates. Statistical difference was analysed using an unpaired t-test; *p < 0.05, **p < 0.001. Columns represent the Mean ± St. Dev of five replicates from two different culture conditions.
III CDKi Modulate the Expression Profile of Key Elements of Cell Cycle and MAPK Signaling Pathway
To interpret the effect of CDKi on cell cycle key molecules, the expression of definite CDKs and cyclins were determined by immunoblotting (Figure 4A). The expression of CDK2, 7 and 9 did not change in CDKi treated cells relative to untreated cells. Besides, purvalanol treatment upregulated CDK1 expression but yet downregulated cyclin B1 expression. Since the contribution of the MAPK signaling on proliferation and tumorigenesis, we checked the expression profile of the key proteins of the pathway and observed a significant decrease in p38 levels by CDKi treatment. In contrast, p-p38 was upregulated in response to Roscovitine and Purvalanol treatment in both wt and Puma deficient cells.
IV CDK Inhibitors Lowers the Polyamine Levels in the Cells and Triggers ROS Generation in HCT116 wt and Puma Deficient Cells
Since we know that polyamine (PA) levels are increased in many cancer cells, we primarily examined the effect of CDKi on PA content of the cells by HPLC analysis. Roscovitine (50 µM) and Purvalanol (30 µM) treatment decreased cellular PA levels in both HCT116 wt (Figure 5A) and HCT116 Puma-/- (Figure 5B) cells. Polyamine metabolism consists of biosynthetic and transport pathway and catabolism pathway. In order to understand the effect of CDKi on the expression of enzymes belonging to these pathways, we performed immunoblotting (Figure 5C). Both Roscovitine and Purvalanol treatment upregulated the Polyamine acetyl oxidase (PAO), a member of the catabolic pathway. As the polyamine catabolism generates toxic H2O2 and reactive oxygen species (ROS), we visualized induction of ROS generation under fluorescent microscope via H2DCFDA staining. As shown in (Figure 5D), CDKi treatment increased ROS generation in wt cells. Exogeneous polyamine treatment enhances the ROS amount induced by CDKi.
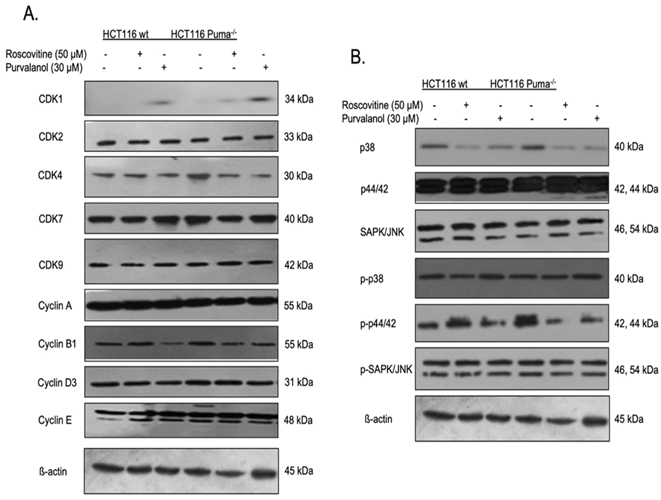
Figure 4: Alterations on CDK, cyclin and MAPK signaling pathway key elements expression profile in CDKi treated HCT116 colon cancer cells. The effect of roscovitine (50 µM) or purvalanol (30 µM) on cycle and MAPK signaling key elements expressions were determined in HCT116 wt and Puma-/- cells by immunoblotting. The expression profile of A) CDK1, 2, 4, 7, 9, cyclin A, B1, D3, E, B) p38, p44/42, SAP/JNK, pp38, pp44/42, ppSAP/JNK was modeled. b-actin was used as a loading control.
V PAO Inhibitor MDL-72527 Reversed CDKi-Induced Apoptotic Cell Death in HCT116 wt and Puma-/- Cells
The reversible effect of PAO inhibitor MDL 72527 on CDKi-induced apoptosis was determined by Cell Death ELISA assay. Results indicate that, although roscovitine treatment induced apoptotic cell death (approximately 4-fold in wt, 2-fold in Puma-/- cells), MDL 72527 co-treatment decreased cell death to about 1,5-fold and 1-fold in wt and Puma-/- cells, respectively (Figure 6A). A similar profile was also observed in Purvalanol-treated cells (Figure 6B). Purvalanol treatment induced apoptotic cell death (approximately 3-fold in wt, 2-fold in Puma-/- cells), whereas MDL 72527 co-treatment decreased cell death to about 1.5-fold and 1.4-fold in wt and Puma-/- cells, respectively. To verify these data, PARP and procaspase-9 cleavage was also examined by immunoblotting. MDL 72527 treatment downregulated CDKi-induced PARP-cleavage in HCT116 colorectal carcinoma cells.(Figure 6C).
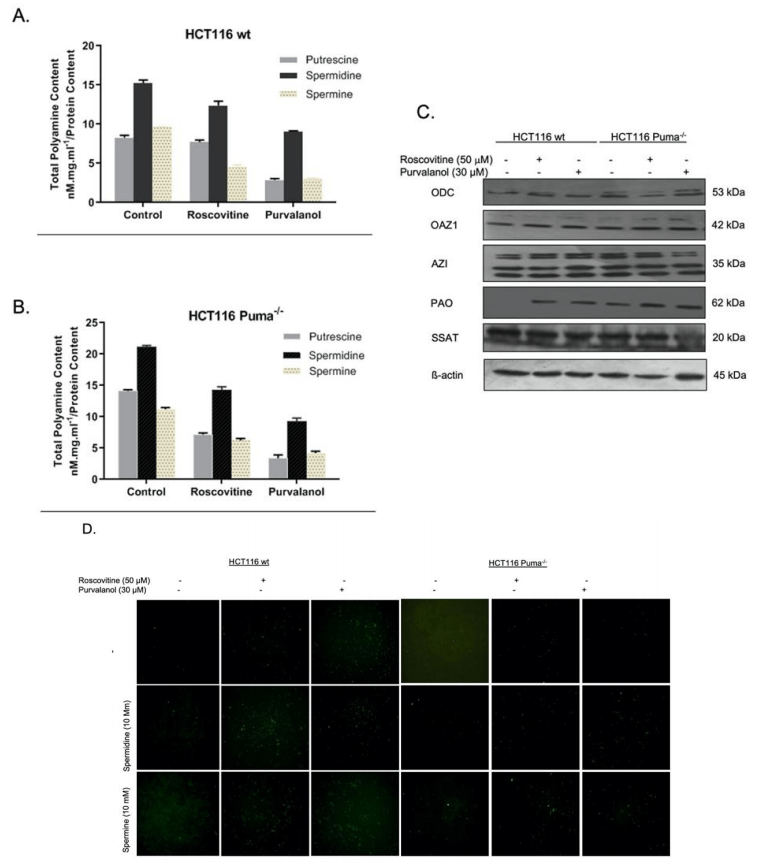
Figure 5: The role of polyamine (PA) metabolism on CDKi-induced apoptotic cell death and ROS generation in HCT116 cells. The effect of Roscovitine or Purvalanol on intracellular PA levels was determined by HPLC analysis in A) HCT116 wt. B) Puma-/- colon cancer cells. C) Expression profiles of ODC, OAZ1, AZI, PAO, SSAT were determined by immunoblotting in HCT116 wt and Puma-/- colon cancer cells following 24 h roscovitine (50 mM) or purvalanol (30 mM) treatments. D) CDKi-induced ROS with/without spermidine or spermine (10 mM each) were determined in HCT116 wt and Puma-/- cells by H2DCFDA (1 µg/ml) staining and visualized by fluorescence microscopy (Magnification was 200x).
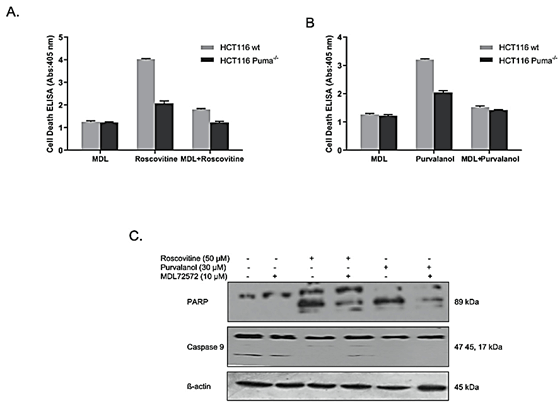
Figure 6: MDL-72527 prevented CDKi-induced apoptotic cell death in colon cancer cells. Reversible effect of PAO enzyme inhibitor, MDL 72527, on A) Roscovitine-induced and B) Purvalanol-induced apoptosis was examined by Cell Death ELISA Assay. Columns represent the means ± S.D. of at least 2 experiments with 4 replicates. Statistical difference was analysed using an unpaired t-test; *p < 0.05, **p < 0.001. C) Immunoblotting method was performed to determine the reversible effect of MDL 72527 on roscovitine or purvalanol-induced PARP cleavage and caspase-9 activation in HCT116 wt colon carcinoma cells.
Discussion
Cell cycle is a complex process, and its regulation involves many proteins such as cyclins and cyclin-dependent kinases (CDKs) [12]. Disruption of signaling and regulation of the cycle may lead to continuous activation of the cycle, which is one of the characteristics of cancer development [13]. Therefore, targeting these regulatory proteins is a promising approach for anti-cancer strategies [5]. Because of their significant roles in cell cycle regulation, several molecules targeting CDKs have been studied. Previous studies show that many CDK inhibitors (CDKi) have been shown to inhibit cell cycle progression and trigger apoptotic cell death in cancer cell lines [14-16]. One of the CDK inhibitors, Roscovitine (CYC202 or seliciclib), is a small purine analog that competes with ATP for binding to targeted CDK and blocks cyclin-CDK complex formation. Recent studies demonstrate that roscovitine induced apoptosis in many cancers such as prostate, breast , leukemia, and non-small cell lung cancer [5, 17-20]. Purvalanol, another purine-derived CDKi, also binds to CDK and prevents its binding to a cyclin and induces apoptosis in different cancer cell lines [21-23]. Accordingly, in this study, we first examined cytotoxic effects of these CDKi in HCT116 wt and Puma-/- cells.
Puma, a BH3- only Bcl-2 family member, is a crucial protein as an initiator of intrinsic apoptosis [24, 25]. Once it is activated, it promotes mitochondrial membrane polarization and induces caspase cascade [25]. It is found that, Puma deficiency reduced drug-induced apoptosis in colon cancer cells [26]. Similarly, both roscovitine and purvalanol lead dose-dependent decrease in cell proliferation and roscovitine increased cell death in wt HCT116 cells (Figures 1A & 1B). Although the decrease of cell viability is not as much as wt cells, CDKi decreased viable cell percentage significantly in Puma-/- cells (Figure 1C). Annexin V-FITC/PI analysis indicates that roscovitine-induced early and late apoptotic cell induction in HCT116 wt cells, whereas Puma-/- cells exerts resistance against the apoptotic induction with regard to wt cells. Strikingly, purvalanol treatment caused late apoptotic and necrotic induction in wt cells in contrast to Puma-/- cells in which apoptosis induction is more significant, which may mean that HCT116 cells are more sensitive to purvalanol than roscovitine and Puma-/- does not affect this sensitivity. As it is known that activation of procaspase-9 by cleavage is the initiator of the caspase cascade in the mitochondrial pathway of apoptosis and subsequently, cleavage of PARP is involved in the pathway [27].
The results verified Annexin V-FITC/PI data in that roscovitine induced cleavage of caspase-9 and PARP, indicating the trigger in apoptosis (Figure 2B). In purvalanol treated cells, although caspase-9 cleavage is not significant as in roscovitine situation, an increase in PARP cleavage by CDKi treatment was obvious (Figure 2C). Previous reports show that well-established drugs and CDKi induce caspase-dependent apoptotic cell death via mitochondrial pathway [5, 6]. It was reported that Puma-/- cells were more resistant than wt cells in drug-induced apoptosis induction [26]. However, our results showed that both CDKi were capable of inducing MMP loss in both wt and Puma-/- cell lines (Figures 3A & 3B). To further examine the caspase-dependency of the CDKi-induced apoptosis, selective caspase inhibitors Z-DEVD-FMK and Z-LEHD-FMK were used, which inhibit caspase-3 and caspase-9, respectively. The caspase inhibitors recovered the CDKi-induced cytotoxicity in HCT116 wt cells. On the contrary, in Puma-/- cells protective effect of caspase inhibitors is not as significant as wt cells.
In this case, cells might induce caspase-independent apoptosis via activation of apoptosis-inducing factor (AIF) and nuclear translocation following mitochondrial activation [28]. When we checked the expression profiles of certain cyclins and CDKs taking part in the cell cycle, immunoblotting results show that both CDK inhibitors upregulated the expression of CDK1 and downregulated the expression of cyclin B1 in both colorectal carcinoma cell lines (Figure 4A). MAP kinase pathways are one of the important cellular pathways in that they modulate essential cellular processes like growth, proliferation, migration upon extracellular signals [29]. They are known to be deregulated in human tumors [30]. While the expression of p38 is downregulated by Roscovitine and Purvalanol treatment, p-p38 is upregulated by these CDKi which suggests the involvement of p38 MAPK pathway upregulation in HCT116 cells (Figure 4B).
Polyamines are small polycationic alkylamines that are involved in many crucial processes of the cell, such as growth, differentiation and tumor growth. Previous studies indicate that intracellular levels of polyamines were increased in cancer cells [10]. Polyamine metabolism is in balance in biosynthesis and catabolism enzymes [11]. Targeting of these enzymes is a promising point in inducing cellular death in cells. The intracellular levels of polyamines decrease in wt and Puma-/- cells following CDKi treatments (Figures 5A & 5B). Moreover, in Puma-/- control cells, polyamine levels were higher than wt cells. However, CDKi treatment also decreased the polyamine levels in Puma-/- cells. After determining the PA levels in response to CDKi, we then checked the expression profiles of the enzymes that are involved in PA metabolism. Although the CDKi had no significant effect on biosynthetic enzymes ODC, OAZ1 and AZI, CDKi treatment significantly upregulated catabolic enzyme, PAO (N-acetyl polyamine oxidase). That means roscovitine and purvalanol induced cell death and decreased the intracellular PA levels via upregulating catabolic activity without any effect on biosynthesis machinery. PAO is one of the essential enzymes in PA catabolism which causes H2O2 generation as a by-product [11]. However, Puma-/- cells did not show the significant effect visualized in wt cells, exogeneous spermidine or spermine treatment induced the ROS generation.
In order to clear up the potential role of PAO in CDKi-induced apoptosis, we used a PAO inhibitor, MDL 72527, along with roscovitine or purvalanol. MDL 72527 is a spermine analog that blocks PA acetylation and decreases H2O2 production, thereby prevents apoptosis. In this study, we examined the effect of MDL 72527 firstly and observed that in both wt and Puma-/- cells, MDL 72527 co-treatment reversed CDKi-induced cell death (Figures 6A & 6B). Also, immunoblotting for PARP cleavage indicated the downregulation of cleaved PARP for both CDK inhibitors.
Conclusion
CDK inhibitors roscovitine and purvalanol induce apoptotic pathways by activation of PA catabolism and after that, decrease of PA levels. Still, alterations of PA catabolism are critical for CDKi-induced cell death.
Author Contributions
ACG: Conception and design, administrative support, data analysis and interpretation, provision of study materials or patients, collection and assembly of data, manuscript writing, final approval of manuscript; BAS: Manuscript writing.
Conflicts of Interest
None.
Article Info
Article Type
Research ArticlePublication history
Received: Wed 07, Jul 2021Accepted: Wed 21, Jul 2021
Published: Wed 04, Aug 2021
Copyright
© 2023 Ajda Coker-Gurkan. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited. Hosting by Science Repository.DOI: 10.31487/j.IJCST.2021.01.03
Figures & Tables
References
1. Rawla P,
Sunkara T, Barsouk A (2019) Epidemiology of colorectal cancer: incidence,
mortality, survival, and risk factors. Prz Gastroenterol 14: 89-103. [Crossref]
2. Nowak
Sliwinska P, Scapozza L, i Altaba AR (2019) Drug repurposing in oncology:
Compounds, pathways, phenotypes and computational approaches for colorectal
cancer. Biochim Biophys Acta Rev Cancer 1871: 434-454. [Crossref]
3. Wang X,
Deng K, Wang C, Li Y, Wang T et al. (2020) Novel CDKs inhibitors for the
treatment of solid tumour by simultaneously regulating the cell cycle and
transcription control. J Enzyme Inhib Med Chem 35: 414-423. [Crossref]
4. Raje N,
Kumar S, Hideshima T, Roccaro A, Ishitsuka K et al. (2005) Seliciclib (CYC202
or R-roscovitine), a small-molecule cyclin-dependent kinase inhibitor, mediates
activity via down-regulation of Mcl-1 in multiple myeloma. Blood 106:
1042-1047. [Crossref]
5. Arisan
ED, Obakan P, Coker Gurkan A, Calcabrini A, Agostinelli E et al. (2014) CDK
inhibitors induce mitochondria-mediated apoptosis through the activation of
polyamine catabolic pathway in LNCaP, DU145 and PC3 prostate cancer cells. Curr
Pharm Des 20: 180-188. [Crossref]
6. Zhang T,
Jiang T, Zhang F, Li C, Zhou YA et al. (2010) Involvement of p21Waf1/Cip1
cleavage during roscovitine-induced apoptosis in non-small cell lung cancer
cells. Oncol Rep 23: 239-245. [Crossref]
7. Arisan
ED, Akkoç Y, Akyüz KG, Kerman EM, Obakan P et al. (2015) Polyamines modulate
the roscovitine-induced cell death switch decision autophagy vs. apoptosis in
MCF-7 and MDA-MB-231 breast cancer cells. Mol Med Rep 11: 4532-4540. [Crossref]
8. Gürkan
AC, Arisan ED, Obakan P, Palavan Ünsal N (2013) Inhibition of polyamine oxidase
prevented cyclin-dependent kinase inhibitor-induced apoptosis in HCT 116 colon
carcinoma cells. Apoptosis 18: 1536-1547. [Crossref]
9. Igarashi
K, Kashiwagi K (2019) The functional role of polyamines in eukaryotic cells. Int
J Biochemist Cell Biol 107: 104-115. [Crossref]
10. Casero
RA Jr., Stewart TM, Pegg AE (2018) Polyamine metabolism and cancer: treatments,
challenges and opportunities. Nat Rev Cancer 18: 681-695. [Crossref]
11. Tracy
RMS, Woster PM, Casero RA Jr (2016) Targeting polyamine metabolism for cancer
therapy and prevention. Biochem J 473: 2937-2953. [Crossref]
12. Wenzel
ES, Singh ATK (2018) Cell-cycle Checkpoints and Aneuploidy on the Path to
Cancer. In Vivo 32: 1-5. [Crossref]
13. Hanahan
D, Weinberg RA (2011) Hallmarks of cancer: the next generation. Cell
144: 646-674. [Crossref]
14. Deng M,
Wang J, Chen Y, Zhang L, Xie G et al. (2016) Silencing cyclin-dependent kinase
inhibitor 3 inhibits the migration of breast cancer cell lines. Mol Med Rep
14: 1523-1530. [Crossref]
15. Yu C,
Cao H, He X, Sun P, Feng Y et al. (2017) Cyclin-dependent kinase
inhibitor 3 (CDKN3) plays a critical role in prostate cancer via regulating
cell cycle and DNA replication signaling. Biomed Pharmacother 96:
1109-1118. [Crossref]
16. Li Y, Ji
S, Fu LY, Jiang T, Wu D et al. (2017) Knockdown of Cyclin-Dependent Kinase
Inhibitor 3 Inhibits Proliferation and Invasion in Human Gastric Cancer Cells. Oncol
Res 25: 721-731. [Crossref]
17. Mohapatra
S, Chu B, Zhao X, Djeu J, Cheng JQ et al. (2009) Apoptosis of metastatic
prostate cancer cells by a combination of cyclin-dependent kinase and AKT
inhibitors. Int J Biochem Cell Biol 41: 595-602. [Crossref]
18. Węsierska
Gądek J, Gritsch D, Zulehner N, Komina O, Maurer M (2011) Roscovitine, a
selective CDK inhibitor, reduces the basal and estrogen-induced phosphorylation
of ER-α in human ER-positive breast cancer cells. J Cell Biochem 112:
761-772. [Crossref]
19. Mohapatra
S, Chu B, Wei S, Djeu J, Epling Burnette PK et al. (2003) Roscovitine inhibits
STAT5 activity and induces apoptosis in the human leukemia virus type
1-transformed cell line MT-2. Cancer Res 63: 8523-8530. [Crossref]
20. Zhang F,
Zhang T, Gu ZP, Zhou YA, Han Y et al. (2008) Enhancement of radiosensitivity by
roscovitine pretreatment in human non-small cell lung cancer A549 cells. J
Radiat Res 49: 541-548. [Crossref]
21. Iizuka
D, Ogura A, Kuwabara M, Inanami O (2008) Purvalanol A induces apoptosis and
downregulation of antiapoptotic proteins through abrogation of phosphorylation
of JAK2/STAT3 and RNA polymerase II. Anticancer Drugs 19: 565-572. [Crossref]
22. Coker
Gürkan A, Arisan ED, Obakan P, Akalın K, Özbe U et al. (2015) Purvalanol
induces endoplasmic reticulum stress-mediated apoptosis and autophagy in a
time-dependent manner in HCT116 colon cancer cells. Oncol Rep 33:
2761-2770. [Crossref]
23. Chen X,
Liao Y, Long D, Yu T, Shen F et al. (2017) The Cdc2/Cdk1 inhibitor, purvalanol
A, enhances the cytotoxic effects of taxol through Op18/stathmin in non-small
cell lung cancer cells in vitro. Int J Mol Med 40: : 235-242. [Crossref]
24. Zheng X,
He K, Zhang L, Yu J (2013) Crizotinib induces PUMA-dependent apoptosis in colon
cancer cells. Mol Cancer Ther 12: 777-786. [Crossref]
25. Chen J,
Zhong J, Liu Y, Huang Y, Luo F et al. (2018) Purified vitexin compound 1, a new
neolignan isolated compound, promotes PUMA-dependent apoptosis in colorectal
cancer. Cancer Med 7: 6158-6169. [Crossref]
26. Chen D,
Wei L, Yu J, Zhang L (2014) Regorafenib inhibits colorectal tumor growth
through PUMA-mediated apoptosis. Clin Cancer Res 20: 3472-3484. [Crossref]
27. Siddiqui
WA, Ahad A, Ahsan H (2015) The mystery of BCL2 family: Bcl-2 proteins and
apoptosis: an update. Arch Toxicol 89: 289-317. [Crossref]
28. Marti BF
(2013) Emerging properties of signaling networks in cancer: a data-derived modeling
approach. Univ Heidelberg.
29. Dhillon AS, Hagan S, Rath O, Kolch W (2007) MAP kinase signalling pathways in cancer. Oncogene. [Crossref]
30. Wagner EF, Nebreda AR (2009) Signal integration by JNK and p38 MAPK pathways in cancer development. Nat Rev Cancer 9: 537-49. [Crossref]