Osteopathia Striata with Cranial Sclerosis Associated with Moyamoya Disease in a 3-Year-Old Girl
Osteopathia Striata with Cranial Sclerosis Associated with Moyamoya Disease in a 3-Year-Old Girl
A B S T R A C T
We present a very rare case of osteopathia striata with cranial sclerosis (OSCS) due to a mutation in the WTX gene, with an unusual association with moyamoya disease (MMD). This is the first documented case to our knowledge where OSCS and MMD have occurred in conjunction. A 3-year-old girl presented with osteopathia striata with cranial sclerosis with a history of Pierre Robin syndrome. She had previously had cleft palate repair and was now electively admitted for an anterior 2/3rds cranial vault remodeling procedure for osteopathia striata. Postoperatively she developed left-sided focal seizures and left-sided weakness. Subsequent imaging revealed acute right frontal and medial right parietal infarcts and bilateral supraclinoid ICA occlusion, consistent with moyamoya disease. Early diagnosis and intervention will alter the prognosis and life expectancy of patients with OSCS and MMD. MMD should be a differential diagnosis following the sudden onset of neurological deficits and focal seizures postoperatively in the pediatric population. In cases where associated comorbidities are high risk, a conservative approach to the treatment of MMD may be indicated.
Keywords
Osteopathia striata with cranial sclerosis, moyamoya disease, WTX gene, Pierre Robin sequence
Introduction
OSCS is a rare X-linked dominant inherited skeletal dysplasia (prevalence 0.1/1 million people) caused by truncating mutations or deletions in the X-linked gene WTX (Wilm’s tumor gene on the X chromosome) and is characterized by sclerotic striations of the metaphyses of long bones, pelvis and scapula along with craniofacial hyperostosis [1, 2]. Germline inactivation of WTX causes OSCS syndrome, which is associated with macrocephaly, frontal bossing, ocular hypertelorism, broad nasal bridge, cleft palate, hearing loss, and mental retardation. Rarely, it also causes cardiac, intestinal, and genitourinary malformations. Moyamoya is a rare progressive, occlusive disease of the cerebral vasculature with particular involvement of the circle of Willis and arteries that feed it, namely the distal internal carotid arteries and the proximal branches of the anterior and middle cerebral arteries [3]. It is the most common etiology of childhood stroke, with the highest incidence in Japan [3]. The female to male ratio is reported as 1.8:1. It is commonly detected by transient ischemic attacks involving motor, speech or sensory functions. Headache, developmental delay, hemiballism, chorea and hemorrhagic stroke have also been reported in the literature as the presenting symptoms [3, 4]. The pathology is described as an initial thickening in the walls of the terminal portions of the ICA vessels bilaterally (intima may show lipid deposits), fibro-cellular thickening of the intima, waving of the elastic lamina and thinning of media.
Case History
Our patient has a significant background history. She was a term birth with a birth weight of 3.5kg, delivered through caesarean section. She is the third child of unrelated parents. Also, of note, she has a brother that has been diagnosed with osteogenesis imperfecta and insulin dependent diabetes mellitus. (Interestingly the brother has a mutation in COL 4A1 [de novo mutation] on Chromosome 7; however, there is no relationship in the genetic conditions between the two siblings). She was diagnosed at 2 months of age with osteopathia striata with cranial sclerosis due to a mutation in the WTX gene (X linked dominant, parental gonadal mosaicism), which almost exclusively affects girls. She also exhibited from birth, low set ears, frontal bossing, and an unusually shaped skull shape. These signs are consistent with OSCS. An initial ECHO demonstrated small ASD and small PDA. She also has Pierre Robin syndrome. Subsequent cleft palate repair was undertaken at 8 months of age. Following that surgery, she developed airway obstruction, and a tracheostomy was inserted. She proceeded to have recurrent respiratory tract infections, and fortunately, she responded well to therapy. She was decannulated 2 years later, and the stoma closed 5 months post decannulation. Insertion of gastrostomy was performed at 10 months of age, and the PEG was reversed 1 year later. She presented to our institution at 3 years of age, with frontal bossing, supraorbital rim flaring, mild hypertelorism, bilateral conductive hearing loss, and developmental delay.
A pre-assessment CT brain for preparation of cranial vault remodeling showed scaphocephaly with sagittal synostosis, premature almost complete fusion of lambdoid suture, and delayed closure of anterior fontanelle. Diffuse thickening of the skull base and calvarium, under pneumatization of the mastoid air cells and paranasal sinuses and bony facial features consistent with the history of osteopathia striata with cranial sclerosis, there is also associated cleft palate, bony defect commencing 5 mm posterior to the incisive foramen (Figure 1A).

Figure 1: A) Preoperative CT brain showing diffuse thickening of the skull base and calvarium. B) Preoperative CT brain showing CT 3D Skull Facial Bones-lateral view. C) Postoperative 3D CT skull showing good approximation.
Operation
After informed consent from the parents, an orbitofrontal distraction procedure, which involved anterior 2/3rds cranial vault remodeling, forehead remodeling with bone graft and parietal widening with osteotomy for osteopathia striata was undertaken. The pericranium was then approximated, and the wound closed in layers. There were no intraoperative complications. Histopathology revealed thickened sclerotic bone. The bone marrow was normocellular, and all three cell lines were present. There was no evidence of malignancy.
Postoperative Care
Day 1 postoperatively she developed left-sided focal seizures (left arm jerking, left facial twitching) 8 seizures were recorded in total lasting approximately between 30s-35mins. The first seizure initially responded to buccal Midazolam, but the seizures were fully abated by Phenytoin and Phenobarbitone. On examination following the seizures, she was noted to have left-sided weakness (left hemiparesis). A subsequent CT Brain revealed acute right frontal and medial right parietal infarcts (Figures 2A & 2B). An MRI brain showed extensive bilateral areas of acute infarct distribution suggestive of watershed infarcts worse on the right side and absence of signal flow within the supraclinoid segment of both internal carotid arteries (ICA). CTA showed absence of flow with the supraclinoid segments of both ICA, in keeping with occlusion and evolution of bilateral infarcts right greater than left (Figure 2C). She was then commenced on Aspirin. IR with general anaesthesia (GA) revealed bilateral supraclinoid ICA occlusion, consistent with moyamoya syndrome (bilateral occlusive cerebrovascular disease) (Figures 3A & 3B). Following the review of imaging by a multidisciplinary team, it was agreed that she was not fit for any pial synangiosis surgery or burr hole surgery in relation to the moyamoya disease as she was at very high risk of further strokes and surgery should be avoided wherever possible as the risk of general anaesthesia would also be considerable for her.
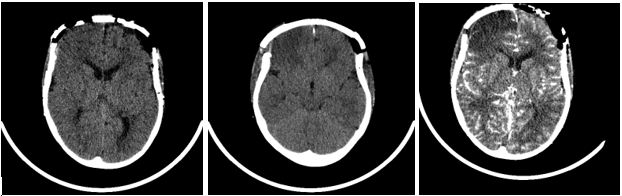
Figure 2: A & B) CT brain shows CT brain right frontal and medial right parietal infarcts. C) CTA shows-Interval expected evolution of the bilateral infarcts (R>L).
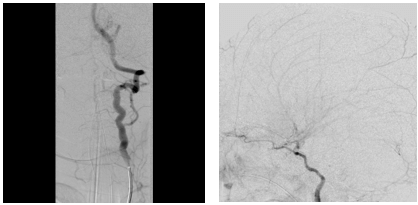
Figure 3: A & B) IR Angio shows: revealed bilateral supraclinoid ICA occlusion, consistent with moyamoya syndrome.
After extensive physiotherapy, on discharge, her head circumference was 59cm > 99percentile, and her weight was 17.6 kg. She also had a mild left upper eyelid ptosis; however, she exhibited full range of motion of her eyes with no VII palsy. The left-sided motor deficit improved with power 3-4/5. She was seizure-free but had an ongoing speech delay which was at the premorbid level. She also complained of mild headaches, which were well managed by analgesia.
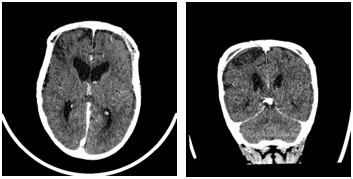
Figure 4: A & B) Non contrast CT brain shows 4A- axial and 4B sagittal views of Left frontal extradural collection, measuring 6 mm in maximum depth, containing a few bubbles of air. This may represent a focus of infection.
She however, returned one week later complaining of seizure like activity, her parents reporting an episode of right arm twitching lasting 10seconds and two episodes of right leg twitching. A subsequent CT brain revealed a left frontal extradural collection, measuring 6 mm in maximum depth containing a few bubbles of air (Figure 4A & 4B). The collection enhanced representing an area of infection. She was started on empiric antibiotic therapy and was later discharged home after a course of antibiotic therapy.
Discussion and Literature Review
OSCS is a rare bone dysplasia characterized by longitudinal long bone striations and cranial sclerosis. Typical facial dysmorphism, sensory defects, internal organ anomalies, growth and mental retardation can also be associated. Osteosclerosis of the cranial and facial bones can lead to disfigurement and disability due to pressure on the cranial nerves [5-7]. OSCS has been associated with a germline mutation in WTX gene (Wilms Tumor in the X, also known as FAM123B and AMER1) [2]. A genetic basis for the phenotypic presentation was proposed by Jenkins et al. They showed that WNT-related signaling defects led to osteoblast activation and inhibition of osteoclast differentiation. WTX knockdown increases transcription of WNT-responsive reporter genes. Genetic analysis of the proband showed a 16-nucleotide deletion c826-841del16 resulting in a frameshift and premature stop codon (WTX gene). In our case, the genetic analysis of the mother was noncontributory.
Moyamoya disease incidence peaks in two age groups, approximately 5 years of age (first decade of life) and adults in 40’s [2]. Females are affected nearly twice as often as males [4, 8, 9]. Patients may present with headaches, seizures, involuntary movements and progressive decline in intellectual ability [10, 11]. Moyamoya disease accounts for 6-14% of all strokes in pediatric patients with conservative management demonstrates poorer intellectual development and proficiency in daily living activities than patients that undergo surgical treatment [8, 9, 12-14]. Some Japanese reports indicate that when untreated, moyamoya syndrome has a poor natural history [15]. The goal of therapy is to arrest or reverse the course of these ischemic events [9]. The antiplatelet effect is useful in moyamoya because some ischemic symptoms appear to occur as a consequence of emboli from micro thrombus formation at sites of arterial stenosis [16, 17]. Smith describes that all patients treated in his study are treated with aspirin indefinitely, however aspirin shows no evidence that it reverses or stops the progression of arterial stenosis [10]. Franco et al. describe two cases that were treated conservatively with aspirin. Motor disabilities improved without any deterioration over a follow-up period of one to four years [18]. In our case, conservative management was ideal considering the extensive history of OSCS and Pierre Robin syndrome in conjunction with MMD.
Conclusion
OSCS and MMD are rare. Early diagnosis and intervention will alter the prognosis and life expectancy of patients with OSCS and MMD. MMD should be in the differential diagnosis following the sudden onset of neurological deficits and focal seizures postoperatively in the pediatric population. In cases where associated comorbidities are high risk, a conservative approach may be indicated.
Article Info
Article Type
Case Report and Review of the LiteraturePublication history
Received: Mon 15, Jun 2020Accepted: Sat 04, Jul 2020
Published: Fri 31, Jul 2020
Copyright
© 2023 Samuel Ndoro. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited. Hosting by Science Repository.DOI: 10.31487/j.SCR.2020.07.06
Figures & Tables
References
- Sean B Herman, Sarah K Holman, Stephen P Robertson, Lynn Davidson, Benjamin Taragin et al. (2013) Severe osteopathia striata with cranial sclerosis in a female case with whole WTX gene deletion. Am J Med Genet A 161A: 649-599. [Crossref]
- Anna Maria Zicari, Luigi Tarani, Daniela Perotti, Laura Papetti, Francesco Nicita et al. (2012) WTX R353X mutation in a family with osteopathia striata and cranial sclerosis (OS-CS): case report and literature review of the disease clinical, genetic and radiological features. Ital J Pediatr 38: 27. [Crossref]
- T Baba, K Houkin, S Kuroda (2008) Novel epidemiological features of Moyamoya disease. J Neurosurg Psychiatry 79: 900-904. [Crossref]
- Nagaraja D, Verma A, Taly M, Abe H (1997) Familial occurrence of Moyamoya disease. Clin Neurol Neurosurg 99: 5162-5167.
- L Kornreich, M Grunebaum, N Ziv, A Shuper, M Mimouni (1998) Osteopathia striata, cranial sclerosis with cleft palate and facial nerve palsy. Eur J Pediatr 147: 101-103. [Crossref]
- Heung Chul Park, Hang Gul Kim, Yong Hwan Kim, Joo Hwan Kim, Moon Young Kim et al. (2014) Osteomyelitis in an Osteopathia Striata with Cranial Sclerosis Patient. Maxillofac Plat Reconstr Surg 36: 285-291. [Crossref]
- Magliulo G et al. (2007) American Journal of Otolaryngology-Head and Neck Medicine and Surgery 28: 59-63.
- Hyunho Choi, Ji Yeoun Lee, Ji Hoon Phi, Seung Ki Kim, Byung Kyu Cho (2013) Postoperative epidural haematoma covering the galeal flap in paediatric patients with Moyamoya disease: Clinical manifestation risk factorsand outcomes. J Neurosurg Pediatr 12: 181-186. [Crossref]
- Edward R Smith, R Michael Scott (2005) Surgical management of Moyamoya Syndrome. Skull Base 15: 15-26. [Crossref]
- Jodi L Smith (2009) Understanding and treating Moyamoya disease in children. Neurosurg Focus 26: E4. [Crossref]
- Tricia S Williams, Robyn Westmacott, Nomazulu Dlamini, Leeor Granite, Peter Dirks et al. (2012) Intellectual ability and the executive function in pediatric moyamoya vasculopathy. Dev Med Child Neurol 54: 30-37. [Crossref]
- T Imaizumi, K Hayashi, K Saito, M Osawa, Y Fukuyama (1998) Long term outcomes of paediatric moyamoya disease monitored to adulthood. Pediatr Neurol 18: 321-325. [Crossref]
- Seung Ki Kim, Kyu Chang Wang, Chang Wan Oh, In One Kim, Dong Soo Lee et al. (2003) Evaluation of cerebral hemodynamics with perfusion MRI in childhood Moyamoya disease. Pediatr Neurosurg 38: 68-75 [Crossref]
- D Nagaraja, A Verma, A B Taly, M V Kumar, P N Jayakumar (1994) Cerebrovascular disease in children. Acta Neurol Scand 90: 251-255. [Crossref]
- R Michael Scott, Jodi L Smith, Richard L Robertson, Joseph R Madsen, Sulpicio G Soriano et al. (2004) Long term outcome in children with moyamoya syndrome after cranial revascularization by pial synangiosis. J Neurosurg 100: 142-149. [Crossref]
- Ohaegbulam C, Scott RM (2001) Moyamoya syndrome. In McLone D, ed. Pediatric Neurosurgery. Philadelphia, PA: WB Saunder 1077-1092.
- R M Scott (2001) Surgery for Moyamoya syndrome? Yes. Arch Neurol 58: 128-129. [Crossref]
- C M Franco, M M Fukujima, R de M de Oliveira, A A Gabbai (1999) Moyamoya disease: report of three cases in Brazilian patients. Arq Neuropsiquiatr 57: 371-376. [Crossref]