Management of Phosgene-Induced Acute Lung Injury (ALI) by Personalized Protective PEEP-ECMO: What Can We Learn from Animal Bioassays?
A B S T R A C T
Background: Phosgene (carbonyl dichloride) gas is an indispensable chemical inter¬mediate used in numerous industrial processes. Acute lung injury (ALI) caused by accidental inhalation exposure to phosgene is characterized pulmonary edema being phenotypically manifested after an asymptomatic or more precisely phrased “clinical occult” period. Opposite to common clinical practice, protective treatment should be given preference to curative treatment. Treatment initiated already during the asymptomatic phase shortly after exposure requires prognostic endpoints preceding the lung edema for triage and re-triage. Treatment strategies need to be personalized and exposure-dose related. The objective of this post-hoc analysis of published data is to assess prognostic value of ventilation dead-space (Vd/Vt) and extravascular lung water index (EVLWI) to guide treatment by protective PEEP supplemented by venovenous (vv) ECMO.
Methods: This paper aims to compare the overarching published framework from systematic toxicological research of phosgene in animal bioassays with the clinical evidence from four accidentally phosgene-poisoned workers admitted to hospital with life-threatening lung edema. Treatment focused on a combination of protective PEEP and ECMO to reverse phosgene-induced deterioration in lung mechanics by personalized mechanical ventilation. Endpoints selected for titration PEEP focused on endpoints indicative of decoupling cardiopulmonary and vascular functions. To better understand any cardiogenic and vascular disturbances, titration endpoints included calculated ventilation dead-space (Vd/Vt), measured extravascular lung water index (EVLWI), arterial blood gases and acid-base status, systemic vascular resistance index (SVRI), and cardiac index (CI). EVLWI and APACHE II criteria guided the course of treatment in adjusting plateau pressure (Pplat), positive end-expiratory pressure (PEEP), and driving pressure (ΔP).
Results: Remarkable equivalence of human data and those from controlled inhalation studies with phosgene on rats and dogs was found. The endpoint of choice guiding PEEP ventilation and implementation of ECMO was EVLWI. This maker of lung edema precisely reflects the increased wet lung weights in animals.
Conclusions: ECMO-supplemented PEEP not only mitigates hypoxemia at conditions of severe ARDS and it also provides a means to reduce driving and plateau pressures minimizing ventilator-associated lung injury.
Keywords
Phosgene, lung irritants, acute lung injury, ARDS, prognostic markers, countermeasures, adverse pathway outcomes, inhalation
Background
Phosgene (carbonyl chloride) is a poorly water-soluble, reactive colorless gas 3.4-times heavier than air at ambient temperature and pressure. Phosgene is an important and indispensable high production volume chemical intermediate in the manufacture of various types of polymeric materials, dyestuffs, pharmaceuticals, and agrochemicals. Commonly, phosgene is manufactured “on demand” from the reaction of carbon monoxide and chlorine gas in the presence of activated charcoal. Depending on the industrial process of synthesis and use, accidental exposure to workers may occur to the gas phase or to organic solvents containing phosgene. Any co-exposure to chlorine is process/accident dependent [1]. Its congeners trichloromethyl chloroformate (diphosgene) and bis(trichloromethyl) carbonate (triphosgene) are liquid and solid, respectively. The median lethal concen-trations (LC50) following 4-hour exposure in rats was 7.2, 13.9, and 41.5 mg/m³ or 1.8, 1.7, and 3.4 ppm for phosgene, di- and triphosgene, respectively. Unlike the acute lung edema-driven mortality patterns observed in phosgene- and diphosgene-exposed rats, triphosgene caused additional delayed mortality patterns [2, 3]. Hence, treatment strategies need to be guided by detailed information on the major irritants involved and the ‘exposure dose’. Two variables must be known, the concentration x exposure time profile and the activity of the exposed subject keeping in mind that the respiration-dependent ‘inhaled dose’ determines the degree of poisoning. Treatment should be guided by triage, prognosis, and toxic-mechanism-related rather than symptomatic treatment.
The avid covalent reactions of phosgene with nucleophilic moieties of directly accessible biomolecules at the portal of entry may cause loss of their functionality and denaturation. Opposite to the more water-soluble reactive irritant gases, phosgene gas is not water-soluble enough to be retained in the airways to any appreciable extent. Thus, the amphiphilic conditions constituted by alveolar surfactant increases its retention at the alveolar level. The ensuing acute lung injury (ALI) is characterized by a decreased dynamic lung compliance due to both dysfunctional surfactant and increasing surface tension at the alveolar air-blood interface [1, 4]. If severe enough, atelectasis follows as determined by the Laplace equation. In accordance with imbalanced hydrostatic and protein oncotic pressures (Starling’s equation), extravasation of fluid from the pulmonary vessels occurs [5, 6]. The fluid and proteins leaking into the airspaces aggravate further dysfunctional pulmonary surfactant. The resultant self-amplifying hetero¬geneous distribution of areas with lower/higher dynamic compliance to dynamic pressures and higher/lower resistance to air flows require pressure- and frequency-adjusted preventive mechanical ventilation to minimize atelectasis of slow and stiff areas and over-distention of the fast and more complaint areas. One of the greatest clinical challenges emerge from the asynchrony of these highly and poorly ventilated areas with differing impedances conducive to ventilation-perfusion disturbances resulting in shunt and ventilation dead-space. Perfusion-related complex cardiogenic and cardiovascular factors may cause redistribution of plasma fluid from the systemic circulation towards the pulmonary circulation.
Related mechanical factors must be appreciated when targeting normoxemia in critically ill patients receiving oxygen. While detrimental effects to the lungs from high levels of oxygen may cause oxidative stress and related toxicity at FIO2 (fraction of inspired oxygen) levels above 0.6-0.7. Especially in lungs with ventilation/perfusion mismatch high inspired oxygen does not necessarily translates to improved oxygenation due to the limited accessibility and capacity of yet functional alveolar units which may attain hemoglobin saturation but normoxemia cannot be attained due to venous admixture. Consistent with published evidence, the following vicious cycle appears to be conceivable: reduced active lung volume and surfactant dysfunction promote small airways closure; this causes alveolar gas trapping; if the trapped gas is mainly oxygen, reabsorption atelectasis will happen rapidly [7, 8]. If high-driving pressure ventilation is used, reabsorption atelectrauma occurring preferentially in the ‘fast’ areas receiving the highest driving pressure. Under such conditions, pneumomediastinum and airway injury is a likely outcome in the small airways that are cyclically opened and closed leading eventually to activation of the inflammatory signaling cascade. Thus, low FIO2 prevents atelectasis.
Regarding early prognostic assessment, hemoconcentration (hypovolemia) was shown to be an early biomarker of the phosgene-induced ALI both in rats and humans [9-11]. Treatment approaches to this finding frequently triggered non-conservative fluid resuscitation and changed a non-lethal to an iatrogenic lethal acute lung edema, as the surplus infusion of fluid was shown to settle in the lung [12, 13]. Hence, fluid resuscitation should be handled by the balancing act of using conservative fluid resuscitation but preventing systemic shock. Unlike the hemoconcentration occurring as a result of small shifts of fluids from the large systemic compartment into the much smaller pulmonary compartment, the measurement of increased extravascular lung water-content (EVLW) appears to be the most direct integrating biomarker of phosgene-induced ALI. Remarkable coherence of the human EVLW scale with the surrogate endpoints increased hemoconcentration and lung weights was shown in phosgene-exposed rats and dog [14-16]. Suffice it to say, despite its lower sensitivity, incremental measurements of hemoconcentration can readily be implemented at the paramedic level, whereas the more specific and prognostic EVLW requires ICU standard. Most of the data regarding medication use in phosgene poisoning are derived either from anecdotal experience in case reports or from studies involving animal models. Case reports are plagued by the absence of a control group and frequently by the lack of any robust documentation regarding level of phosgene exposure as gas, liquid (pure or dissolved in a liquid) or gas/solid in case of triphosgene. Although considered “global technical standard”, readings from medical phosgene badges often are not reported. Animal studies have been instrumental for elucidating pathophysiological mechanisms and providing initial measures of treatment by drugs. Pharmacological treatment strategies of human phosgene-induced ALI have not found their way into the clinic yet; however, studies using preventive mechanical ventilation to buy time for self-repair and reconstitution of lung function appeared to be efficacious both in larger animals (dogs and pigs) and humans. The objective of this paper is to review and compare the conclusions from published evidence on animal bioassays with the PEEP/ECMO treatment strategy applied at the Nanchang Hospital on accidentally exposed workers to potentially lethal doses of phosgene.
Methods
I Patients
Thirteen factory workers were exposed to phosgene gas at a chemical plant on March 31, 2018. Within 1~2 hours after this incident workers were admitted to a local hospital for triage and treatment. One day post-incident, four workers reached a critical condition evidenced by symptoms of pulmonary edema and were transferred to the ICU of this hospital. All four patients were male; within the age of 34~45 years (average: 46.25±8.58 years) and were reported to be in good health with no special medical history. Pulse oxygen saturation (SpO2) was about 0.85. The acute physiology and chronic health evaluation II (APACHE II) scores within 24h after admission to the ICU were 24-29 (average: 26.50±2.08) which meets the diagnostic criteria of severe acute phosgene poisoning andacute respiratory distress syndrome (ARDS) [17]. Shortly after admission at ICU (day 0) the four patients were intubated and subjected to positive end-expiratory pressure mechanical ventilation (PEEP). Patients were relative refractory to oxygen inhalation which prompted ECMO-supplemented personalized PEEP treatment within 24-hours post-admission to ICU. Large amount of edema fluid was drained from the accessible airways by suction. Chest CT examinations showed that both lungs were "ground glass-like" with patchy infiltration and patchy consolidation. One patient had complications with subcutaneous and mediastinal emphysema and right pneumothorax. Body temperatures were normal, pulse was between 100 to 120 beats/min; respiration was between 28 to 35 breaths/min, and the blood pressures were in the range 135-150/65-78 mmHg. Patients were conscious, with shortness of breath. Auscultation of the lungs revealed crepitant rales, but rhonchi or wheezes may also be present. Electrocardiogram suggested sinus rhythm. Chest X-ray suggested diffuse patchy shadows in both lungs (Figure 1). Clinical pathology revealed normal liver and kidney function, hematology provided evidence of increased neutrophils and hemoglobin. Arterial blood gas analysis revealed hypoxemia, hypercapnia, and hyperlactemia [17]. Phosgene is produced from carbon monoxide and the airway irritant gas chlorine, production-related incidences cannot exclude co-exposure to chlorine as well. Unlike the phosgene-induced ALI causing alveolar injury and edema with rapid recovery, the chlorine-induced ALI is characterized by a more insidious characteristics of injury and lingering inflammation of airways. If the acute edema phase of phosgene poisoning is survived, delayed mortality is not uncommon in chlorine-exposed animals and humans as a result of obliterating bronchiolitis. Although shown to be ineffective for phosgene poisoning, corticoids were demonstrated to be effective for mitigating the chlorine-induce ALI [18-20]. As an exact exposure history was not available to the clinicians, corticoids were administered as a precautionary approach.

Figure 1: The four patients’ Chest x-ray on April 1. Chest x-ray posteroanterior view shows diffuse patchy shadows in both lungs and the heart shadow size is normal.
II Treatment
i General treatment and procedures
The patients’ health status was controlled by the following measurements: ECG, arterial blood gas and acid-base analyses, serum lactic acid, hematology, and hemostasis. Blood was collected from a subclavian venous catheter and femoral artery catheter. Hemodynamics, including EVLWI: pulse-indicated continuous cardiac output (PiCCO) monitoring (for details see below). Supportive treatments: deep sedation and analgesia, antibiotics for preventing pulmonary infection, and titration anti-coagulation using Heparin (or details see ECMO). Heparin infusion was applied to maintain activated clotting time (ACT) at 140-160 sec at an infusion rate of 2.5 to 3.6 mL/min. Hence, appropriate conservative fluid resuscitation was targeted; the cumulative fluid balance (CFB) was conservative (≤ 80 mL fluid/kg/day). Methylprednisolone 80 mg i.v. infusion 2x/day with gradual reduction of dose during the course of treatment, bronchodilator, norepinephrine to maintain blood pressure, propofol, and atracurium as muscle relaxant.
ii Ventilator settings
The ventilator settings applied after admission to ICU (day 0) and during ECMO-supplementation on days 1, 3, and 7 are detailed in Results and Discussion. Due to the deteriorating status of patients’ refractory to oxygen supply, PEEP ventilation started on day 0 with invasive ventilation with volume control ventilation (VCV) mode through endotracheal intubation. Setting of ventilator parameters before ECMO treatment: ventilation frequency of 20~22 times/min, TV of 6~8 mL/kg, PEEP 10 to 15 cmH2O (1 cmH2O = 0.098 kPa). Driving pressures (ΔP) were monitored during ECMO treatment, with adjusted ΔP, adjusted TV to 3~6 mL/kg, ventilation frequency to 10 times/min, PEEP to 8 to 10 cmH2O, and inhaled oxygen concentration (FiO2) of0.30. After the improvement of conditions (day 7), ECMO supplementation was discontinued and the VCV mode of ventilation was changed to the pressure supported ventilation (PSV) mode. Extubation was followed by oxygen supply via mask.
iii Venovenous (VV)-ECMO cannulation
During VV-ECMO the blood is drained into the circuit, where it is oxygenated and carbon dioxide is removed, following which it is returned to the patient's venous system. This configuration relies on the patient's own circulation and requires adequate cardiac function. Cannulation: femoral vein - centrifugal pump - membrane lung - internal jugular vein. Heparin was continuously infused to maintain the active coagulation time (ACT) in the range of 140 to 160 sec and the flow rate at 2.5~3.6 L/min. The blood temperature was maintained at 36.5 to 37.0 ˚C using temperature-controlled jacketed cannulas.
iv Feed-back parameters for ventilator setting
Arterial blood pH, partial pressure of carbon dioxide (PaCO2), partial pressure of oxygen (PaO2), blood lactic acid (Lac) and systemic vascular resistance index (SVRI), cardiac index (CI), extravascular lung water index (EVLWI), plateau pressure (Pplat), PEEP, ΔP, and APACHE II, treatment duration of ECMO, duration of mechanical ventilation and duration of ICU stay before and 1, 3 and 7 days after ECMO treatment. Although PaO2 is on important symptomatic endpoint, the measurement of excess EVLW was taken as an integrating marker of improvement in lung mechanics. Incremental measurements of the body weight adjusted EVLW (EVLWI) served as basis for re-adjusting the personalized ventilator settings to the improvement of lung mechanics. Increase in pulmonary vascular permeability accompanied with accumulation of excess EVLWI was reported to be the hallmark of ARDS [15, 16]. Currently, EVLWI and pulmonary vascular permeability index (PVPI) can be quantitatively measured using the transpulmonary thermodilution (TPTD) technique. Clinical standards for hemodynamic and EVLWI measurements have been applied: PiCCO monitoring system [ProAQT platform or PiCCO2 monitoring (Pulsion/Getinge Medical Systems, Munich, Germany)] was used. This system requires a central venous catheter and a thermistor-tipped arterial catheter. After injection of 15 ml of cold isotonic saline into the central venous catheter, the arterial catheter detects thermodilutional changes, which allow for estimation of cardiac output, including the EVLW.
III Animal Reference Studies
i Rats
Excised lung weights were determined post-mortem at the climax of lung edema 1-day post-exposure to phosgene. Lung weights were determined prior to bronchoalveolar lavage (BAL) for analysis of total protein and soluble collagen. Collagen proteins in BAL were thought to better differentiate lower and higher molecular weight proteins since the former may bypass the vascular-alveolar barrier function more readily. Due to the procedures used in experimental animal studies (exsanguination in terminal anesthesia), lung weights were normalized to concurrent air-exposed control groups. Six rats/group were nose-only exposed in restraining tubes for 30- or 240-min to multiple concentration x time (C x t) relationships ranging from the non-toxic to the time-adjusted non-lethal concentration (LCt01). In rats, the time-adjusted acute non-lethal threshold concentration (LCt01) and median lethal concentration (LCt50) were 1075 and 1741 mg/m³ x min, respectively [1, 14]. This matrix of data was complemented by BAL protein data from a published study on rats acutely exposed for 6-hours ranging from air only (control) to 1476 mg/m³ x min from Hatch et al.) and analyzed for BAL protein postexposure day 1 [21].
ii Dogs
In a proof-of-concept study, three groups of four Beagle dogs each were head-only exposed once for 30 minutes to phosgene gas under otherwise identical conditions to those applied for rats. Apart from rats, dogs were placed in a sling with minimum restraint. However, unlike rats, dogs do not show the nociceptive stimulations typical for small rodents. These include but are not restricted to hypothermia and changes in cardiopulmonary reflexes. Due to the narrow lumen of the airways of small rodents’, reflex-mediated protective proteinaceous secretions into airways occur. Thus, increased BAL protein due to loss in alveolar barrier function is superimposed by secreted proteins from the airways. Such confounding factors do not occur in dogs (nor in humans). The excised lung was weighed and the lobus accessorius was lavaged similar to rats. The dog study used actual exposure concentrations and Cxts of 0, 9, 16.5, and 35 mg/m³ equivalent to 0, 270, 495, and 1050 mg/m³ x min, respectively. Sham control dogs were used as reference [1, 14]. At the point of departure (POD), the transition from non-adverse to clinically significant outcomes, at which the increased EVLWI meets an increasingly blocked lymphatic drainage, a foudroyant flooding of the alveolar space can be expected. In the normal human lung, the EVLW reflects the equilibrium between fluid leakage and lymphatic drainage [14-16, 22-24]. For reference, an EVLWI value of 7.3±2.8 mL/kg body weight for the normal population. An EVLWI from patients presenting 9 mL/kg, the lung edema was judged “borderline”, whereas EVLWI values above 10 mL/kg were reported to represent higher than normal EVLWI [25]. An EVLWI above 15 mL/kg bw was concluded to be the criterion for severe pulmonary edema. Controlled inhalation studies with phosgene on rats and dogs revealed that the “severe” score in humans equals the non-lethal threshold POD (LCt01) in rats [16, 14]. For the human-to-rat/dog comparisons made in this paper, the PODs of increased EVLWI of 10 and 15 mL/kg were taken to scale bioassay data to the respective EVLWIs from patients on the admission day. Normalization was applied for the human EVLWIs resulting in 100, 137 and ≈200% at 7.3, 10, and 15 mL/kg, respectively. The same normalization was applied for the wet lung weights from rat and dogs exposed to phosgene [14].
IV Ventilation Dead-space
Studies on rats exposed to phosgene revealed a marked drop of the expired CO2 occurring already during the early asymptomatic phase post-exposure [14, 10]. This qualifies CO2 to be an early prognostic marker preceding the phosgene-induced acute lung edema. It could be speculated that an early surge of vasoconstrictive cytokines leaking from dysfunctional apoptotic/necrotic alveolar macrophages may have caused reduced perfusion of yet functional alveolar areas resulting in a higher retention of vascular CO2 at the expense of its reduced exhalation [26-30]. Depending on the disease condition, this secondary etiopathology can contribute to an elevated physiological dead space (Vd) and increase in overall ventilation-perfusion disturbances caused by ventilation delivered to non-perfused alveolar units. At clinical settings, the measurement of Vd/Vt received heightened recognition as a prognostic biomarker of ALI and proved to be a powerful predictor of mortality in patients with ALI/ARDS and was identified as means for optimal PEEP adjustment. Vd/Vt was calculated in patients as suggested by Siddiki and coworkers [26]. Patients subjected to PEEP without ECMO the Vd/Vt was in the range of 0.5; a similar ratio was also observed in phosgene-exposed rats [10]. However, calculations of ventilation dead-space were confounded by ECMO due to the enhanced clearance of CO2 in the extracorporeal circuit rather than in the pulmonary circulation. Therefore, ventilator adjustments on EVLWI was given preference.
V Statistical Analysis
SPSS 19.0 software was used for data analysis. The measurement data are presented as mean ± standard deviation. A one-way ANOVA was used for comparison before and after self-treatment; and Dunnett-t test was used for pairwise comparison. P < 0.05 was considered a statistically significant difference to the feed-back endpoints measured on day 0.
Results and Discussion
I Phosgene-induced ALI - clinical significance of EVLW
The pathophysiological hallmark of phosgene-induced ALI is characterized by the accumulation of fluid in the alveoli and interstitium, characterized as excess EVLW. Remarkable coherence of the human EVLW scale with the surrogate endpoint ‘increased lung weight’ was shown in phosgene-exposed rats and dog (Figure 2). The relative elevations in lung weights were indistinguishable up to an EVLWI of 137% corresponding to 10 mL/kg body weight in humans (Figure 2). The POD towards a beginning clinically significant lung edema evidenced by increased lung weight was ≈600 mg/m³ x min. The POD (LCt01) in rats for lethality was 1072 mg/m³ x min and matched favorably for dogs as well. The higher variability of dogs was attributed to hyperventilation (panting); those dogs with higher ventilation presented more severe poisoning. Similar observations were made by other authors who found that the individual dog’s minute ventilation correlates significantly with the changes observed post-exposure to phosgene [31].
Accounting for the species-specific differences in respiration of rats, dogs, and humans under non-stressed conditions, the body weight-adjusted respiratory minute ventilation (MV/kg-min) is 0.8, 0.4, and 0.2 L/kg-min, respectively [32]. The relationship shown in (Figure 2) suggests that hyperventilating dogs had a MV twice that of normally breathing dogs. For the extrapolation of increased lung weights toward the mean EVLWI from the four patients on the admission day, the relationship from rats is given preference to that from dogs because of the larger data base covering the range from non-lethal to potentially lethal. From the relationships given in (Figure 2), be it from rats or of the hyperventilating dogs, the Cxt or inhaled dose of workers can be estimated 1170 mg/m³ x min. However, when assuming MVs of workers during the accident in the range of 0.2 to 0.3 L/kg-min, these workers may have inhaled an about 4- to 2.7-fold lower volume than rats under this given mode of exposure and activity. Consequently, in order to attain inhaled dose-EVLWI-equivalence, workers were most likely exposed to concentrations of phosgene in the range of 3200 to 4500 mg/m³ x min. This calculated integrated inhaled dose-effect relationship is not at variance with that given by the factory which estimated exposure to phosgene gas at 100-300 mg/m³ for 15 to 30 minutes. This translates to about 4500 mg/m³ x min. As detailed below, such an exposure intensity appears to be plausible from an entirely clinical perspective. Biomarkers in BAL commonly are believed to mirror pulmonary edema. This supposition was verified/refuted in rats and dogs acutely exposed to phosgene. Opposite to the matching relationships of increased EVLW and lung weights (Figure 2), elevations in total protein in BAL were conspicuously more pronounced in rats compared to dogs (Figure 3).
One would have expected that the extravasation of high molecular weight collagen is more specific than the determination of total proteins in BAL. The results from rats show than an appreciable amount of proteins probed by BAL appear unrelated to the Cxt-related increased lung weights depicted in (Figure 2). It seems as if rodent-specific nociceptive reflexes serving protection, increase protein secretion into the airways. Similar reflexes are not operative in the more human-like dog (Figure 3). This comparison corroborates this notion that EVLW-based feed-back markers for personalized treatment strategies counteracting pulmonary edema serve the purpose best. Thus, a note of caution should be sounded concerning the use of analyses from sputum; although readily available, the source and direct interrelationship of selected biomarkers to pulmonary edema remain a conundrum. Accordingly, although the treatment strategy described in this paper was guided by both hemodynamic and physiological variables, particular attention was directed towards continuous re-adjustment of mechanical ventilation guided by EVLWI. Calculated ventilation dead-space was considered but proofed to be not applicable as detailed in methods.
II Translational Dosimetry
The human non-lethal threshold dose of phosgene (LCt01) and median lethal exposure dose (LCt50) are reported to be 1200 and 2000 mg/m³ x min, respectively [33]. More recent published evidence considers 1500 mg/m³ x min as the LCt50 in the healthy adult human population [34]. These hallmarks of phosgene-induced pulmonary toxicity and the estimated inhaled dose calculated above anticipate an early occurrence of pulmonary edema within a short asymptomatic period. This prediction coincides with the condition of patients admitted to ICU presenting progressive dyspnea, cyanosis, a large amount of moist rale in both lungs, protein-rich sputum and tracheal secretions. From that combined evidence it is concluded that the phosgene-exposed workers inhaled a dose of phosgene in the range 3-times the human LCt50. Information from any worn “medical phosgene badge”, a mandatory recommendation of the global phosgene-producing industry, was not available. Dosimeter readings in the range and above 1200 mg/m³ x min require rapid admission to specialized ICU centers. However, it needs to be recalled, such gas-diffusion device does not breathe, i.e. the inhaled dose from heavily breathing workers is subject to underestimation. At variance with the often-applied symptomatic treatment strategy, workers exposed to such levels of phosgene must undergo treatment well ahead before clinical evidence of potentially lethal lung edema is apparent. Notably, any clinically manifest pulmonary edema becomes increasingly refractory to any treatment.
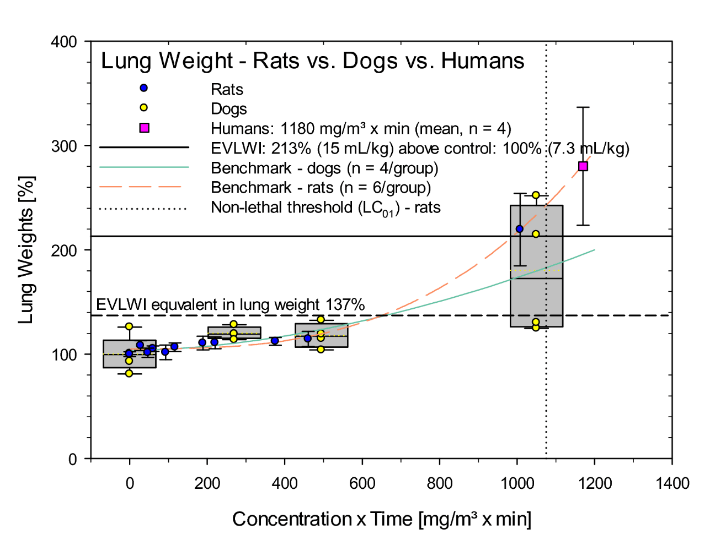
Figure 2: Comparison of lung weights from rats exposed nose-only to a range of Cxt products of phosgene for 30- or 240-min. Dogs were exposed head-only for 30 min to 0, 9, 16.5 or 35 mg phosgene/m³. Lung weights were determined 1-day post-phosgene exposure. Data points represent meansSD (rats: n = 6, dogs: n = 4). Data were normalized to the air control group (= 100%). The curve and the benchmark doses (BMD) were calculated from the data shown and fitted to the polynomial model according to US-EPA [35, 36]. The lower and upper boundaries of the boxes represent the 25th and 75th percentiles, respectively. Medians and means are represented by solid and doted lines, respectively. Data points represent individual dogs. The mean EVLWI was from patients described in the paper (Day 0).
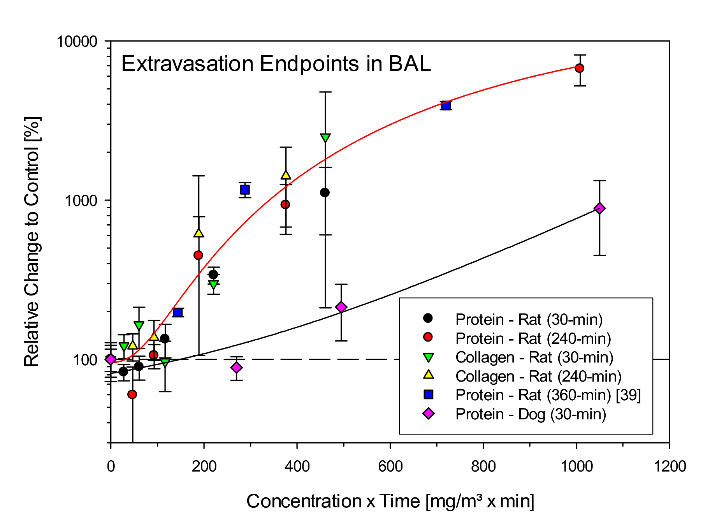
Figure 3: Comparison of the biomarkers of extravasation in bronchoalveolar lavage (BAL) total protein and collagen from rats or dogs exposed by inhalation to Cxt products of phosgene for 30, 240 and 360 min (data reproduced from Pauluhn 2006b and Hatch et al., 2001). Total protein and soluble collagen were determined 1-day post-phosgene exposure. Data points represent meansSD (rats: n = 6; dogs: n = 4). Data were normalized to the air control group (= 100%).
III Personalized treatment by VV-ECMO-supplemented PEEP
Fluid resuscitation was targeted to be conservative to prevent any superimposed aggravation of lung edema due to dysregulated nervous control of the cardiopulmonary system [14]. Published evidence suggests that fluid overload by infusion may precipitately aggravate lung edema. Ventilator settings were continuously monitored and re-adjusted to the improvement of oxygenation and EVLW as summarized in (Figure 4 and Figure 5). Initial PEEP was ≈15 cmH2O with hypoxemia difficult to improve. Low Vt ventilation was targeted at Vt of 4-6 mL/kg with patients in prone position did not improve the patients’ condition. However, with the onset of using VV- ECMO on day 1 precipitous improvement occurred (Figure 4 and Figure 5). The respective ventilator settings and degree of improvement are detailed in (Table 1). ECMO-supplementation not only improved oxygenation (which can be expected), it also improved cardiovascular and pulmonary mechanics integrated by decreasing EVLWI. This outcome demonstrates that the EVLWI guided adjustment of ventilator settings with reduction in driving and plateau pressures was suited improve the phosgene-induced ALI without VILI-related side effects.
Collectively, all four patients presented with the phosgene-specific characteristics of the acute respiratory distress syndrome (ARDS) were successfully treated by VV-ECMO-supported PEEP observing a stepwise titration of pressures to EVLWI as index of reduction of lung edema and improved lung mechanics. Considering that the average initial EVLWI met the score “greater than severe” - which translates to a definite lethal outcome - turned to precipitous improvement by using VV-ECMO-supported titration PEEP in the absence of VILI. The duration of using ECMO was 6-12 days (average 8.0±2.7 days) whereas that of mechanical ventilation was 6-20 days (average 10.75±6.19 days). All 4 patients were discharged after 6-20 ECMO days and 8-27 ICU days. After discharge, delayed adverse findings were reported.
Notably, past treatment strategies focused on pharmacologic and symptomatic interventions with emphasis to improve hypoxia rather than the underlying deteriorating lung mechanics and cardiopulmonary disturbances typically occurring at high EVLWIs. However, anti-inflammatory pharma¬cological principles must be considered when mitigating injuries caused by airway irritants, such as chlorine. Recalling that any increased EVLWI interrelates with decreases lung compliance and increased elastic recoil of lung tissues. Functional corollaries of these changes are mechanically compressed pulmonary airways and vessels. Recalling that common bronchodilators target to activate beta-2 receptors, the bronchial smooth muscle of the airways relaxes but the degree of tissue-related compression remains unchanged. Accordingly, the premise of successful treatment requires mechanistic knowledge on the mode to action of toxic gases as well as the inhaled dose involved. From any clinical perspective, the most integrated course of treatment is to interrupt the vicious cycle leading to increased EVLWI by preventing or reversing the deterioration in lung mechanics rather than using symptomatic interventions. In summary, the phosgene-induced pulmonary edema is caused by factors seriously affecting two basic laws of lung mechanics and function. It seems logical that an acute lung injury caused by mechanical derailments can be treated best by titrated mechanical measures counteracting the underlying mechanical dysfunction. In other words, “similia similibus curantur” still appears to be a preferred approach.
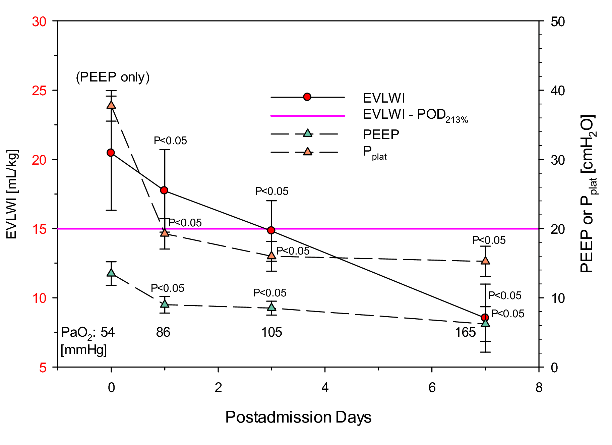
Figure 4: Time-course data of ventilator settings compared with results from determinations of from partial pressures of arterial oxygen in blood. EVLWI 15 mL/kg is the point of departure (POD) scored at “clinically severe lung edema”. When normalized to the normal EVLWI of 7.3 mL/kg (100%) the severe score becomes ≈200%.
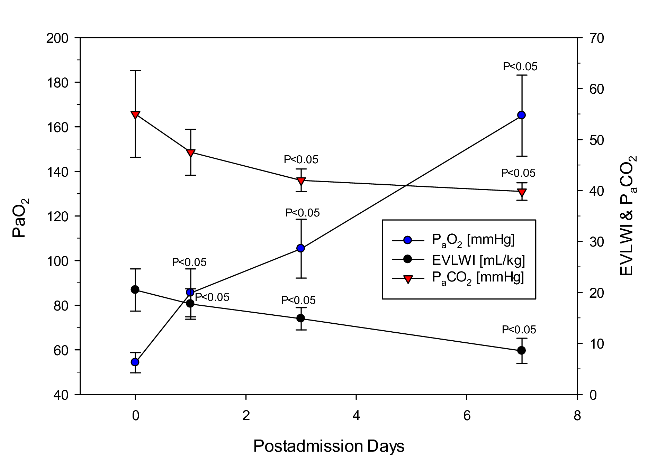
Figure 5: Time-course data of comparing with results from determinations of from partial pressures of arterial oxygen and CO2 in blood with that of EVLWI.
Conclusions
The application of VV-ECMO in Chinese hospitals has made tremendous progress. This mode of ECMO is mainly applied to severe ARDS patients since oxygenation and carbon dioxide removal is achieved by gas exchange in the extracorporeal circuits rather than the lungs. ECMO supplemented PEEP supports hypoxemic conditions which cannot be improved even by extreme PEEP known to cause ventilator related lung injury. We found that vasoactive drugs can be spared to maintain the patients’ hemodynamics stable. Knowing that phosgene-induced ALI occurs parallel to exposure by a clinically occult onset of disturbed fluid-balance of the lung, Vd/Vt determined by volumetric capnometry seems to be invaluable early prognostic marker for triage in asymptomatic patients. Ideally treatment should be preventive and not curative, i.e., being initiated already during the asymptomatic phase. In the moribund patients described in this paper, EVLWI is shown to be the titration marker of choice for combined PEEP-ECMO. Respiratory mechanics and hemodynamics must be observed for pressure titration. Overall, integrated clinical wisdom is needed to understand the modes of action of any inhaled toxic gas and the physiological sequels that may follow progressively. Successful interruption of vicious cycles cannot be achieved by “trial and error” approaches. Rather, disease-specific personalized countermeasures with feed-back control for improvement of health should be given preference.
Availability of Data and Materials
The datasets used for this study are available from the corresponding author on request.
Acknowledgement
We thank Prof. Dr. Juergen Pauluhn, Covestro Deutschland AG, Global Phosgene Steering Group, 51365 Leverkusen, Germany, for sharing original data from previously published papers which allowed us to directly compare data measured in human patients accidentally exposed to phosgene and treated in our hospital with similar or surrogate data from phosgene-exposed rats and dogs.
Funding
There is no financial interest or any third-party involvement that would have influenced the interpretations given in this paper. There are no conflicts of interests. No funding was available for this research.
Contributions
HZ was one of the attending physicians during the patients’ hospital stay, performed data collection and management and drafted the manuscript; JP contributed to the manuscript regarding aspects of translational dosimetry and toxicology. YX was one of the attending physicians during the patients’ hospital stay, surveying the course of treatment, analyzed and interpreted patient data. YC was one of the attending physicians during the patients’ hospital stay and was the principal contributor to this paper. All authors read, revised, and approved the paper.
Ethics Approval and Consent to Participate
This study meets the criteria of medical ethics and is approved by the Hospital Ethics Committee (approval number: 2018051). All patients’ family members granted signed informed consent.
Consent for Publication
Not applicable.
Conflicts of interest
The authors declare that they have no competing interests.
Availability of Data and Material
Not applicable.
Abbreviations
ACT: activated clotting time
ALI: Acute Lung Injury
APACHE II: acute physiology and chronic health evaluation
ARDS: Acute respiratory distress syndrome
CFB: cumulative fluid balance
CI: cardiac index
CO2:Carbon dioxide
ΔP: driving pressure
ECG: electrocardiography
ECMO: extracorporeal membrane oxygenation
EVLWI: extravascular lung water index
FRC: functional residual capacity
FiO2: fraction of inspired oxygen
LC01: non-lethal threshold concentrations
LCt01: time-adjusted non-lethal threshold concentration
LC50: median lethal concentration
LC50: time-adjusted median lethal concentration
MV: respiratory minute volume
Palv: the pressure at the alveolus
Pao: the pressure at the airway opening
PaO2: partial pressure of oxygen in arterial blood
PaCO2: partial pressure of carbon dioxide in arterial blood
PEEP: positive end-expiratory pressure
PL: transpulmonary pressure
POD: point of departure
Pplat: plateau pressure
SVRI: systemic vascular resistance index
TLC: Total lung capacity
V/Q: ventilation/perfusion
vv: venovenous
Vd/Vt: ventilation dead-space fraction
Vd:physiological dead space
Vt: tidal volume
