Large Retroperitoneal Perivascular Epithelioid Cell Neoplasm (PEComa): A Case Report and a Brief Review
A B S T R A C T
Objective: To describe a case of retroperitoneal perivascular epithelioid cell tumor (PEComa) and to discuss the main features of this rare pathology.
Introduction: PEComas represent a rare cluster of neoplasms with uncertain origin; their precursor cells are spindle-shaped and characterized by a myomelanocytic phenotype, so only immunohistochemical staining makes a definitive diagnosis possible. To date, less than three hundred cases are reported in Literature and retroperitoneal site accounts for 7-8% of overall locations.
Case Report: Middle-aged female has visited for abdominal pain and urinary complaints; physical findings and imaging demonstrated a huge inhomogeneous mass occupying right abdomen and arising from renal capsule. After multidisciplinary evaluation, patient has been addressed to open surgery and an en-bloc resection of the mass, with right nephrectomy and adrenalectomy. Immunohistochemical staining made a diagnosis of PEComa possible. After an uneventful postoperative stay, the patient entered a follow up protocol, without signs of local recurrence and distant metastases.
Conclusion: Retroperitoneal PEComa often presents as a bulky mass with renal and adrenal involvement. Surgical resection should be aimed to obtain a complete removal with negative margins; this makes compartment surgery and en-bloc resection mandatory. Immunostaining is the key methods for a correct diagnosis.
Keywords
PEComa, soft tissue tumor, perivascular epithelioid cell, angiomyolipoma, clear cell sugar tumor, lymphangiomyomatosis
Introduction
Perivascular Epithelioid Cell tumors (known as PEComas) represent rare mesenchymal tumors; the first PEComa was described by Apitz in 1943; since then, only case reports and few small series are reported in the literature [1]. This tumor can occur in various sites of the human body: the most common are uterus (20%), skin (8.2%), liver/falciform ligament (8%), retroperitoneum (7.7%) and colon/rectum (6.8%); additional cases affect lung, bone, heart, nasopharynx and omentum [2]. The precursor cell of PEComa is currently unknown: normally no perivascular epithelioid cells exist, but they typically stain both for melanocytic markers (Human Melanoma Black-45 - HMB45-, Melan A, microphthalmia transcription factor -Mitf-) and myogenic markers (actin, myosin, calponin) [3]. This feature links PEComa to other neoplasms, such as angiomyolipoma (AML), lymphangioleiomyomatosis (LAM) and clear-cell sugar tumor (CCST) while, genetically, PEComa often expresses the Tuberous Sclerosis genes TSC1 and TSC2; this evidences led the World Health Organization (WHO) to recognize in its renewed 2002-classification of soft-tissue tumors a family of neoplasms showing similar morphologic and immunohistochemical features but different biological behaviour: size, mitotic rate, infiltrative growth and vascular invasion have been advocated to assess potential malignancy [4]. Whenever arising into retroperitoneal space, PEComa often exhibits a slow but steady growth; in this site PEComa must be distinguished from clear cell sarcoma, smooth muscle neoplasm, gastrointestinal stromal tumor (GIST), adipocytic tumor and adrenocortical carcinoma, but a preoperative definition is difficult, and diagnosis can be reached only after pathological examination and immunohistochemical staining [5]. As a consequence of this attitude, tumor is usually bulky at diagnosis, with a clear aspect of renal and adrenal infiltration and an open multiple organ resection is required in order to obtain a complete surgical removal. The case of the young woman we recently observed and successfully treated for a giant renal PEComa led us to reconsider clinical and surgical aspects related to this rare pathology.
Case Report
A 42-year-old female from Philippines was referred to our department through the emergency ward unit of our hospital on August 2020 for abdominal pain, hematuria and stranguria. Neither previous abdominal surgery nor significant familiarity was referred, but only 2 full term pregnancies; no sweating, weight loss or increasing weakness were reported. Patient complained moderate to severe pain on the right flank and hypochondrium (5.5 points in the Pain Scale Chart); a large mass could be clearly detected on the upper right quadrant at physical examination. Laboratory tests revealed Hb 14.6 g/dl; WBC 10 x 103 uL and RPC 4.06 mgr/dl. A 11.5 x 7.8 x 11 cm solid expansive mass with density ranging from 15 to 50 Hounsfield units, with areas of striae and nodulations was visualized at emergency abdominal CT scan without contrast; tumor displaced the lower margin of right hepatic lobe, the hepatic colonic flexure and the upper pole of the right kidney. Adrenal gland could not be identified with certainty. These findings suggested the presence of a voluminous renal angiomyolipoma or, as alternatives, an adrenal mass or a retroperitoneal liposarcoma. A panel of tumoral markers were tested on admission with the following results: CA 125: 9.9 U/mL; Ca 15.3: 12.6 U/mL; CA 19.9: 3 U/mL; CEA: 1.49 ng/mL; alpha FP: 1.78 ng/mL; other blood tests confirmed absence of anemia and coagulation was within range.

Figure 1: A) Contrast CT scan (arterial phase) A large inhomogeneous mass occupying right abdomen arises from right renal capsule without any cleavage (arrow); B) Intra-tumoral artero-venous shunt (arrow); C) Contrast CT scan (venous phase) Evidence of multiple large pericapsular veins (arrows); D) Evidence of large retroperitoneal venous circulation draining into right renal vein (arrows).
Contrast CT scan did not show cranium or chest nodularities; the right kidney appeared under-rotated due to the presence of a bulky formation with adipose and inhomogeneous density (from 20 Hounsfield Unit on pre-contrast images to 75 Hounsfield on post-contrast image), arising from the capsule of the upper pole of the right kidney (which was clearly infiltrated) as an exophytic growth (Figure 1A), measuring 18 cm in maximum diameter and imprinting the right hepatic lobe, the gallbladder and the second portion of the duodenum. Some hyperdense striae and hypervascularized areas were observed within the mass. A secondary branch of the renal artery to the upper pole also supplied tumoral mass with intralesional arterio-venous shunts (Figure 1B). A collateral perirenal venous circulation was visualized, particularly on the tumor capsule (Figure 1C) and in the retroperitoneal area, discharging into the right renal vein (Figure 1D). No significant abdominal lymphadenopaties were detected. The multidisciplinary oncologic board authorized a direct surgical approach, without further investigation or neoadjuvant therapies. Size of the mass, which clearly precluded tumor enucleation and partial nephrectomy, its diffuse perirenal venous circulation (Figure 1C) and possibility of a consistent post-embolization syndrome contraindicated both CT-guided biopsy and a selective preoperative arterial embolization. The patient was scored as ASA 2 at anaesthesiological preoperative assessment; volume of the mass and its vascular supply made a postoperative ICU stay advisable.
Surgery was performed with the patient laying on left lateral position, through a Makuuchi incision (reverse “L” incision). The presence of a large mass almost occupying the whole right retroperitoneum with clear aspects of venous hypervascularization was confirmed. Tumor was indissociable from the right kidney, the right adrenal gland and the gallbladder. After right parietocolic dissection and extensive Kocher's maneuver, the right anterolateral aspect of the inferior vena cava (IVC) was dissected free up to the caudate lobe and the right adrenal gland was visualized. The right ureter was sectioned between clips and the right gonadic vein was dissected and followed up to its ending into IVC. Then, the right kidney with the bulky mass were mobilized cranially. The presence of numerous enlarged venous vessels arising from the capsule of the tumor and draining into the posterior aspect of the right renal vein was confirmed. The right renal artery was divided between clips while venous efferences and the renal vein were treated with 45 mm. vascular stapler and 4/0 polypropylene running sutures. After fundus first gallbladder dissection and section of the right adrenal vein, the tumor was removed. The resected specimen measured cm 22 x 16 and weighed gr. 1,325 (Figure 2).
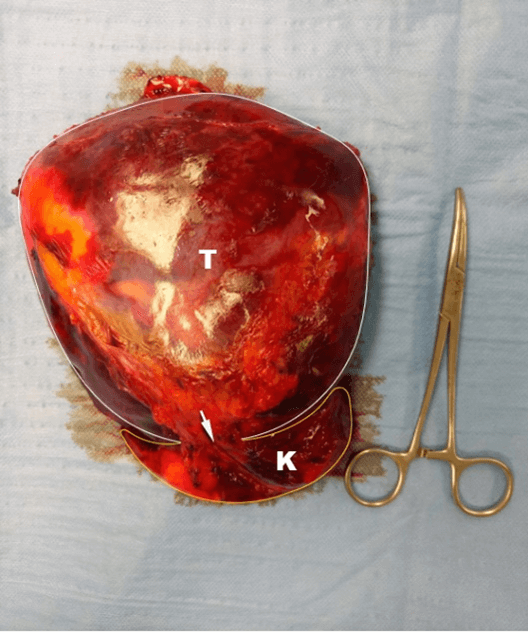
Figure 2: Resected specimen Evidence of encapsulated cm 22 x 16 mass (Tumor) in continuity with right renal capsule (Kidney); absence of any cleavage is confirmed (arrow).
Estimated blood loss was 325 ml.; no blood transfusion was required. The postoperative course was uneventful; after 2 days of ICU stay, patient was transferred to our ward. Drainage was removed on postoperative day 3. Bowel movement started on postoperative day 4 and the patient was discharged on postoperative day 7. At the macroscopic pathological examination, lesion presented as a yellow to gray well circumscribed mass, with a firm and fleshy cut surface; focal hemorrhage and extensive necrosis could be detected. The tumor infiltrated the right adrenal gland clearly and the renal capsule and parenchyma focally (Figure 2). Histology revealed a neoplasm consisting of a combined proliferation of adipose tissue and smooth muscle elements, with an evident vascular component, the latter represented by vessels of different sizes, with frequent hyalinosis of the walls (Figure 3). Around the vascular walls, there were spindle cells arranged in a circumferential orientation, with eosinophilic cytoplasm and atypical nuclei, sometimes pleomorphic. Rare mitosis (1x50 HPF) and areas of tumor necrosis were found. No figures of angioinvasion or aspects of clear infiltration of renal vein and ureter were described. At immunohistochemical staining the spindle cells showed the following phenotypical findings: Actin SM +, HMB45 + (Figures 4A & 4B), Vimentin+, S100-, CD31-, CD34-, CKAE1/AE3-; growth fraction (Ki 67 index) was <3%. The complex of morphological and immunophenotypic findings (in particular, the double expression of Actin SM and HMB45) supported the diagnosis of PEComa (Perivascular Epithelioid Cell tumor). According to Folpe criteria, prognostic factors were conflicting size of the neoplasm, greater than 5 cm, infiltrative growth pattern towards the renal parenchyma, presence of tumor necrosis and, in some areas, the high nuclear grade, indicated a possible disease progression, while rare mitosis, absence of angioinvasion and low Ki67 index suggested a better outcome [6]. The patient entered a close follow up programme at our oncological outpatient clinic with no symptom or sign of disease recurrence at 10 months since surgical treatment.
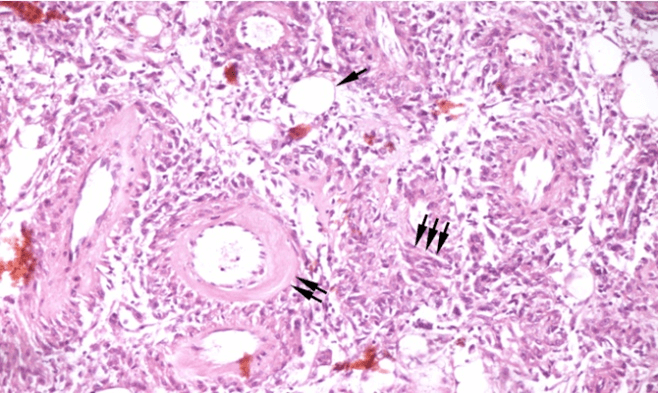
Figure 3: Microscopical evidence for the three tissue components: mature adipose tissue (↑), thick-walled blood vessels (↑↑) and spindle cells arranged in irregular sheets (↑↑↑).

Figure 4: A) Tumoral smooth cells component reacts both for actin and B) melanoma-associated antigen HBM-45.
Discussion
PEComas are a rare cluster of tumors that can arise from any location in the body; in an English Literature review and multivariate analysis, 234 cases of PEComa were described up to 2012 [2]. To date, less than one hundred additional cases have been reported in more recent Literature [7-15]. These tumors originate from perivascular epithelioid cells, which cannot be described and stained in normal tissues; these cells may be encountered in tumors of different locations and biological behaviour: the PEC tumor family include renal AML, CCST of the lung and LAM; other similar rare tumors arising from soft tissue and bones can be defined as PEComa “not otherwise specified” [16]. Association with tuberous sclerosis is also reported: this autosomal dominant syndrome results from mutations and loss of heterozygosity from one of the two genes, TSC1 and TSC2: these patients often develop renal masses, mostly AML, but also malignant PEComa are reported [5].
PEComas are commonly seen in women during the fourth-fifth decade of life: in Bleeker’s review, the median age was 43 years, whereas 79% of patients were female; they are usually asymptomatic but may cause abdominal pain once they grow to a large size [2]. Some patients develop symptoms from external compression and displacement of surrounding organ, whereas general complaints (sweating, fatigue, weight loss, anemia) are extremely rare [17]. Preoperative diagnosis is very difficult, because they may mimic features of many other tumors and the only pathognomonic feature is the co-expression of muscle and melanocytic markers [16]. Immunohistochemistry plays a crucial role in avoiding misdiagnosis of PEComa: they stain for HMB-45, Melan-A, SMA, vimentin; they may be positive for desmin, CD-31, CD-34. They are negative for CgA (chromogranin A), Syn (synaptophysin), CK (creatine kinase), CD-117, CD-10, AFP (alpha fetoprotein) and EMA (epithelial membrane antigen) [18, 19]. Contrast ultrasonography, contrast-enhanced computed tomography and magnetic resonance imaging have been proposed for the imaging assessment of PEComa. At present, no univocal diagnostic algorithm has been proposed [20]. Imaging usually reveals a regular mass, with well-defined borders and maximum diameter ranging from 4-22 cm; lesion is significantly and heterogeneously enhanced on arterial phase, less enhanced on portal venous phase and slightly hypodense on delayed phase [20, 21]. The enhancement pattern varies because of proportion of different components, such as adipose tissue, blood vessels and smooth muscle cells [16]. Tumor frequently appears as partially solid and partially cystic, and displays moderate and disomogeneous contrast uptake [8, 22].
Biological behaviour and malignant attitude of PEComas is still a matter of debate: some PEComas exhibit malignant features, whereas others can be labeled as having uncertain malignant potential or clearly benign. Folpe et al. in 2005 proposed several prognostic factors to categorize any PEComa as follow: benign, malignant or uncertain malignant potential; they include: tumor size > 5 cm, high nuclear grade, hypercellularity, mitotic rate > 1/50 HPF, presence/absence of necrosis, infiltrative attitude at peripheral edges and presence/absence of vascular invasion (Table 1) [4]. If the tumor has none of this, it is considered benign; if it meets less than two criteria, malignancy is uncertain; otherwise, if there are 2 or more prognostic indicators, the tumor is malignant. According to a more recent revaluation, the overall malignant attitude of PEComa has decreased from 71% to 51%: primary tumor size ≥ 5 cm (p= 0.02) and mitotic rate (p < 0.0001) were the only factors significantly associated with recurrence following surgical resection at multivariate analysis [2]. Malignant PEComas are aggressive tumors and may metastasize early, whereas few cases of local recurrences and late distant metastases are reported [7, 12, 16, 22]. Patients with uncertain or benign PEComas have to be followed closely as well, because the natural history of this tumor still needs to be clearly defined [16].
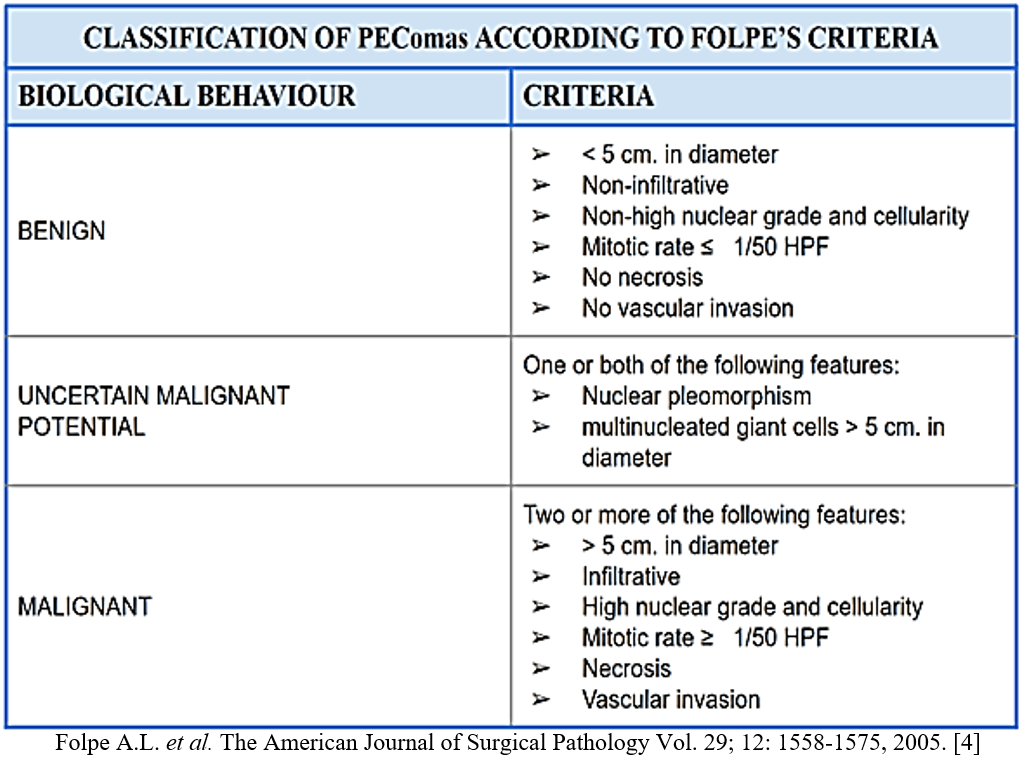
Table 1: Classification of PEComas according to Folpe’s criteria.
Surgery is the only definitive treatment, and an open approach is usual; some form of surgical treatment or excisional biopsy is possible in 92% of patients [2]. Surgical removal should always be aimed to obtain a complete resection of the neoplasm with negative margins at histology examination [23]. Mass large size, infiltrative growth and absence of well-defined boundaries may address to multi-visceral resection, particularly when retroperitoneal location occurs: in these cases tumor removal is frequently combined with radical nephrectomy, adrenalectomy, ureteral resection and vescical wedge resection [7, 11]. Whenever PEComas arise in other locations (ovary, uterus, great omentum), they can be managed easily and sometimes with a video-assisted approach [8, 14]. Concerning renal and retroperitoneal PEComas, preoperative transarterial embolization has been advocated in order to decrease tumor size, intraoperative blood loss and risk of life-threatening hemorrhage; however, experiences are limited to renal AML, whenever a partial nephrectomy could be achieved. Furthermore, the incidence of post-embolization syndrome is high (80-85% rate). Therefore, in malignant and/or giant PEComas, that mandates radical nephrectomy, the benefits of preoperative endovascular treatment still need to be proved [24-26]. There is no consensus regarding the possible role of neo-adjuvant/adjuvant therapy in patients with malignant PEComa: an ultimate diagnosis is not possible until after surgical resection in most. Patients treated by neoadjuvant therapy are limited and chemotherapy appears more useful in the treatment of local recurrences and distant metastases [2, 23]. Several lines of systemic therapies have been proposed (doxorubicin/ifosfamide, paclitaxel/carboplatin, vinorelbine, 5-flurouracil/calcium leucovorine) but always with mild to poor response; targeted therapies with Sunitinib (50 mg/day, 4 weeks) have been proposed for treating abdominal recurrences in patients operated for malignant renal PEComas. Promising results have been shown by the mTOR inhibitor everolimus (10 mg/day): partial response and delay in disease progression for 36 months have been reported in metastatic retroperitoneal PEComa [2, 22, 23, 26, 27].
The case reported confirms principles of diagnosis, treatment and need for a close pathology assessment of large retroperitoneal PEComas. Contrast enhanced CT is the imaging technique of choice for the evaluation of tumor vascularization and relationships. Compartment surgery is mandatory for en-bloc removal of the mass, even if adjacent organs are not clearly infiltrated. Immunohistochemical staining is the key method for a correct diagnosis and addressing the patient to possible adjuvant treatments and optimal follow-up.
Conflicts of Interest
None.
Funding
None.
Article Info
Article Type
Case Report and Review of the LiteraturePublication history
Received: Tue 22, Jun 2021Accepted: Mon 05, Jul 2021
Published: Mon 19, Jul 2021
Copyright
© 2023 Giorgio Lucandri. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited. Hosting by Science Repository.DOI: 10.31487/j.SCR.2021.07.10
Figures & Tables
References
1.
Apitz K (1943) Die Geschwülste und
Gewebsmissbildungen der Nierenrinde. II Midteilung. Die mesenchymalen
Neubildungen. Virchows Arch 311: 306-327.
2.
Bleeker JS, Quevedo JF, Folpe AL (2012) "Malignant"
perivascular epithelioid cell neoplasm: risk stratification and treatment
strategies. Sarcoma 2012: 541626. [Crossref]
3.
Hornick JL, Fletcher CDM (2006) PEComa: what do we know so
far? Histopathology 48: 75-82. [Crossref]
4.
Folpe AL, Mentzel T, Lehr
HA, Fisher C, Balzer BL et al. (2005) Perivascular epithelioid cell neoplasms of soft tissue
and gynecologic origin: a clinicopathologic study of 26 cases and review of the
literature. Am J Surg Pathol 29: 1558-1575. [Crossref]
5.
Martignoni G, Pea M, Reghellin D, Zamboni G, Bonetti F (2008)
PEComas: the past, the present and the future. Virchows Arch 452:
119-132. [Crossref]
6.
Folpe AL, Kwiatkowski DJ (2010) Perivascular epithelioid cell
neoplasms: pathology and pathogenesis. Hum Pathol 41: 1-15.
[Crossref]
7.
D’Andrea D, Hanspeter E, D’Elia C, Martini T, Pycha A (2016)
Malignant Perivascular Epithelioid Cell Neoplasm (PEComa) of the Pelvis: A Case
Report. Urol Case Rep 6: 36-38. [Crossref]
8.
Okamoto K, Okada Y, Ohno K, Yagi T, Tsukamoto M et al. (2018)
A rare case of perivascular epithelioid cell tumor (PEComa) of the greater
omentum. World J Surg Oncol 16: 113. [Crossref]
9.
Liegl B, Hornick JL, Fletcher CDM (2008) Primary cutaneous
PEComa: distinctive clear cell lesions of skin. Am J Surg Pathol 32:
608-614. [Crossref]
10.
Yamashita K, Fletcher CDM (2010) PEComa presenting in bone:
clinicopathologic analysis of 6 cases and literature review. Am J Surg
Pathol 34: 1622-1629. [Crossref]
11.
Sukov WR, Cheville JC, Amin MB, Gupta R, Folpe AL (2009)
Perivascular epithelioid cell tumor (PEComa) of the urinary bladder: report of
3 cases and review of the literature. Am J Surg Pathol 33: 304-308. [Crossref]
12.
Bennett JA, Braga AC, Pinto A, Van de Vijver K, Cornejo
K et al. (2018) Uterine PEComas: A Morphologic, Immunohistochemical, and
Molecular Analysis of 32 Tumors. Am J Surg Pathol 42: 1370-1383. [Crossref]
13.
Zhao J, Teng H, Zhao R, Ding W, Yu K et al. (2019) Malignant
perivascular epithelioid cell tumor of the lung synchronous with a primary
adenocarcinoma: one case report and review of the literature. BMC Cancer
19: 235. [Crossref]
14.
Sánchez Gálvez M, Parra Membrives P,
Sánchez Bernal ML, Martínez Baena D, Lorente Herce JM et al. (2020) Hepatic
PEcoma: an unusual tumor in an infrequent location. Cir Cir 88: 215-218.
[Crossref]
15.
Westaby JD, Magdy N, Fisher C, El Bahrawy M (2017) Primary
Ovarian Malignant PEComa: A Case Report. Int J Gynecol Pathol 36:
400-404. [Crossref]
16.
Vijayabhaskar R, Mehta SS, Deodhar KK, Pramesh CS, Mistry RC
(2010) PEComa of the lung. J Cancer Res Ther 6: 109-111. [Crossref]
17.
Abhirup B, Kaushal K, Sanket M, Ganesh N (2015) Malignant
hepatic perivascular epithelioid cell tumor (PEComa) - Case report and a brief
review. J Egypt Natl Canc Inst 27: 239-242. [Crossref]
18.
Danilewicz M, Strzelczyk JM, Wagrowska Danilewicz M (2017)
Perirenal perivascular epithelioid cell tumor (PEComa) coexisting with other
malignancies: a case report. Pol J Pathol 68: 92-95. [Crossref]
19.
Tay ShY, Lao WT, Chen ChL, Chan WP (2013) Contrast-enhanced
ct and angiographic findings in hepatic perivascular epithelioid cell tumor. JBR-BTR
96: 308-310. [Crossref]
20.
Chang H, Jung W, Kang Y, Jung WY (2012) Pigmented
perivascular epithelioid cell tumor (PEComa) of the kidney: a case report and
review of the literature. Korean J Pathol 46: 499-502. [Crossref]
21.
Tirumani SH, Shinagare AB, Hargreaves J, Jagannathan JP,
Hornick JL et al. (2014) Imaging features of primary and metastatic malignant
perivascular epithelioid cell tumors. AJR Am J Roentgenol 202: 252-258. [Crossref]
22.
Tan Y, Xiao E (2012) Hepatic perivascular epithelioid cell
tumor (PEComa): dynamic CT, MRI, ultrasonography, and pathologic
features--analysis of 7 cases and review of the literature. Abdom Imaging
37: 781-787. [Crossref]
23.
Hulová S, Sycova Mila Z, Macák D, Janega P, Chovanec M et al.
(2018) Dia-gnostic Challenges and Extraordinary Treatment Response in Rare
Malignant PEComa Tumor of the Kidney. Klin Onkol 31: 448-452. [Crossref]
24.
Otegi Altolagirre I, de
Miguel Valencia M, Sánchez Acedo P, Zazpe Ripa C, Tarifa Castilla A et al. (2017) Our experience
in the surgical treatment of liver PEComa. Gastroenterol Hepatol 40:
24-28. [Crossref]
25. Camps V, Maertens V, Michiels M, Plasschaert H,
Ceulemans R (2021) Perirenal perivascular epithelioid cell tumor (PEComa). Acta
Chir Belg 17: 1-4. [Crossref]
26. Wang D, Li HZ, Ji ZG (2017) Effectiveness and safety of laparoscopic enucleation combined with selective arterial embolization for renal angiomyolipoma. Cancer Biomark 19: 177-183. [Crossref]
27. Flum AS, Hamoui N, Said MA, Yang XJ, Casalino DD et al. (2016) Update on the Diagnosis and Management of Renal Angiomyolipoma. J Urol 195: 834-846. [Crossref]