Large Orbital Tumor in a Patient with Neurofibromatosis Type 2
A B S T R A C T
Neurofibromatosis type 2 (NF2) is a separate entity from Neurofibromatosis type 1 (NF1) or von Recklinghausen’s disease, and is much less frequent than NF1. Vestibular schwannomas are the hallmark lesion, affecting 95% of individuals and typically occur bilaterally. Schwannomas commonly occur on other nerves intracranially and in the spinal compartment, along with meningiomas, ependymomas, and gliomas. Ophthalmologic abnormalities are present in the majority of NF2 patients, including cataracts, retinal changes, optic nerve sheath meningiomas and other optic pathway tumors. These tumors are sometimes large and can induce significant proptosis. We report the case of a large orbital meningioma responsible for severe proptosis in a patient with NF2.
Keywords
Neurofibromatosis, type 2, tumor, schwannoma, meningioma, proptosis
Introduction
Neurofibromatosis 2 (NF2) is an autosomal dominant disease whose hallmark is the development of bilateral vestibular schwannomas (VS). Schwannomas also occur on the other cranial, spinal and peripheral nerves. Other manifestations are intracranial, spinal and optic nerve sheath meningiomas. We report the case of a large orbital meningioma responsible for severe painful proptosis, optic nerf compression and complete visual loss in a patient with NF2.
Case Report
A 16-year-old girl, followed for neurofibromatosis type 2 since years, she presented with severe painful proptosis and chemosis of her left eye. She had pre-existing cognitive disorders, hearing loss and imbalance related to bilateral vestibula schwannoma. She had one-year history of worsening headaches and proptosis. The clinical examination found, on the left eye, an important non-axial proptosis, with significant chemosis (Figure 1). There was no light perception, no pupil light reflex, eye movements were restricted, and the optic disc was atrophic.
MRI revealed multiples lesions that are well seen with contrast-enhanced T1-weighted MRI. T2-weighted MRI sequences showed lesions with peritumoral edema.
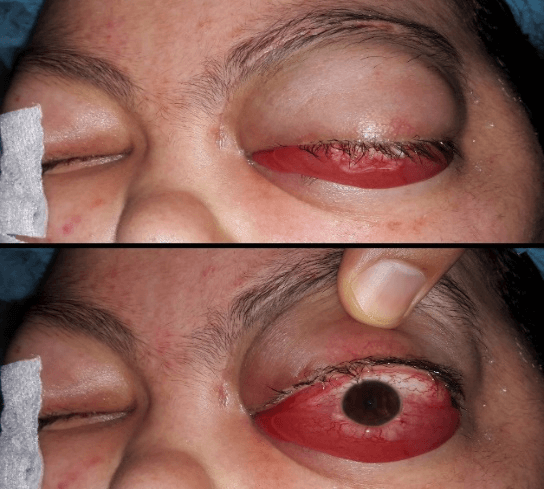
Figure 1: Important proptosis of the left eye, with significant chemosis.
All these tumors had the same signal characteristics: left orbital lesion measuring 50x20mm including the optic nerve with extension to the internal extraconal space and responsible for a proptosis grade 2; at the left cerebellopontine angle measuring 20x15mm, and the right angle measuring 10mm, and in front of the olfactory bulb. This MRI appearance suggested neurofibromatosis affecting the left optic nerve, olfactory nerve and bilateral acoustic nerves (Figure 2).
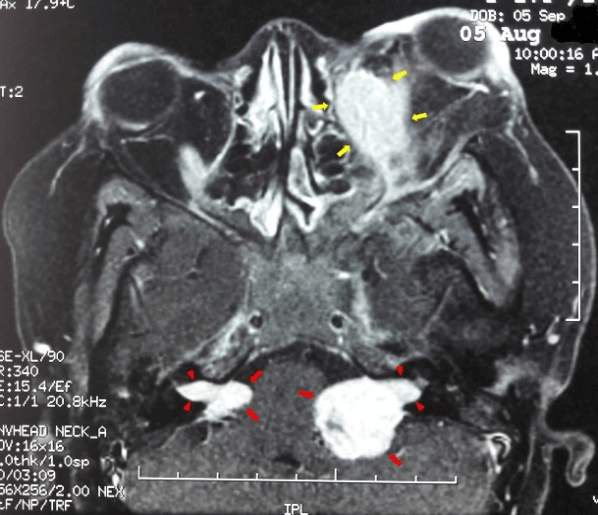
Figure 2: Axial T2-weighted contrast-enhanced MRI reveal, left orbital meningioma (yellow arrows) including the optic nerve with extension to the internal extraconal space and responsible for a proptosis grade 2, and bilateral vestibular schwannomas (red arrows). Intracanalicular extension (arrowheads) of tumors into the internal auditory meatus is clearly delineated.
The patient underwent tumor resection by an internal conjunctival approach (Figure 3), which made it possible to remove, in fragments (Figure 4), a large part of the orbital tumor mass. The resection was incomplete due to the size of the tumor, its posterior extension and the lack of individualizable limits. The pathology study confirmed the diagnosis of meningioma..
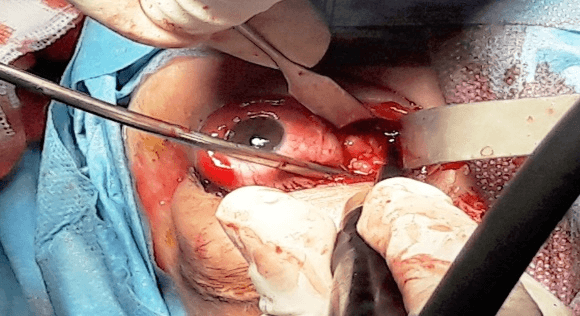
Figure 3: Surgical excision by an internal conjunctival approach.
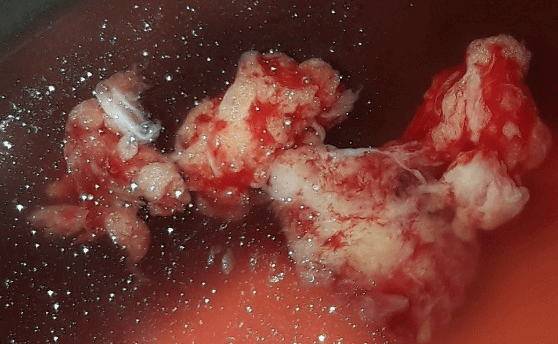
Figure 4: Some resected fragments of the orbital tumor.
Discussion
Neurofibromatosis type 2 is an autosomal dominant, characterized by multiple nonmalignant nervous system tumors, including schwannomas, meningiomas, ependymomas and gliomas, with the cardinal feature being bilateral vestibular schwannomas. Incidence is estimated at 1 in 33 000-40 000 and prevalence 1 in 100 000 [1].
An abnormality can be identified in the NF2 gene. It is a tumor suppressor gene located on chromosome band 22q12 [1]. This gene encodes a protein known as merlin (a moesin-ezrin-radixin-like protein) or schwannomin [2]. Merlin is important in anchorage of the cytoskeleton to the cell membrane, the organization of cell membrane proteins and interaction with cytosolic proteins. The pathways involved are required for cell growth, protein translation and cellular proliferation. The absence of normal merlin is associated with a predisposition to tumor formation [3]. Although NF2 can be inherited from an affected individual, the majority of patients acquire de novo mutations in the NF2 gene [4]. It has been shown that approximately 25-33% of the de novo cases are mosaic, with somatic mosaicism being defined as the presence of genetically distinct populations of somatic cells in a single organism [5].
Table 1: Manchester criteria for clinical diagnosis of NF2.
|
|
Additional findings needed for diagnosis |
|
Bilateral vestibular schwannomas |
None |
|
1st degree family relative with NF2 |
Unilateral vestibular schwannoma or two NF2-associated lesions (meningioma, glioma, neurofibroma, schwannoma, or cataract) |
|
Unilateral vestibular schwannoma |
Two NF2-associated lesions associated with the disorder (meningioma, glioma, neurofibroma, schwannoma, or cataract) |
|
Multiple meningiomas |
Unilateral vestibular schwannoma or two other NF2-associated lesions (glioma, neurofibromas, schwannoma, or cataract) |
NF2 is typically diagnosed at age 20-30 years, but features are often present for many years before the diagnosis is made [6]. About 10% of patients present before the age of 10 years, and 18% before the age of 15 years, with a greater diversity of clinical features than seen in adults, and often with increased severity [7]. The majority of patients experiencing hearing loss as their presenting symptom. Loss of balance is one of the major problems encountered as the disease progresses, with eventual death due to the various complications occurring as a result of the tumor load. Within families, the clinical manifestations of NF2 are often very similar, however there can be a large variation in symptoms between non-related individuals [7]. Ophthalmologic abnormalities are present in the majority of NF2 patients, including cataracts (70-80%), retinal changes (20-44%), strabismus (12-50%), amblyopia (12%), optic nerve sheath meningiomas and other optic pathway tumors (10-27%), and extra-ocular movement abnormalities (10%) [8-11]. Nystagmus may occur due to peripheral vestibular dysfunction or eye involvement. Corneal injury affects about 10% of NF2 patients with other ocular abnormalities like proptosis and in association with facial nerve weakness. The diagnosis of NF2 is based only on clinical criteria (Table 1), vestibular schwannomas are the hallmark lesion, typically occur bilaterally [12].
Despite its benign histological nature, intracranial tumors in NF2 are associated with significant morbidity; they can grow and exert a pressure effect onto important surrounding anatomical structures leading to cortical blindness, hearing loss, gait abnormalities, increased intracranial pressures, paralysis, seizures and even death. Vital anatomical structures, contained within the cranial vault, can often make surgical resection of these tumors complex and challenging [13]. Unfortunately, visual impairment is not uncommon in NF2. All patients with orbital meningioma require close follow-up with serial MRI, visual field, and acuity testing. Successful strategies for preserving vision in NF2 patients with orbital meningioma include observation, microsurgical resection of tumor, or optic nerve decompression with or without radiosurgery [14]. Surgical excision is mainly discussed for symptomatic lesions. In our case, given the disfiguring orbital location and the large size of the tumor with compression of the optic nerve and complete visual loss, we opted for tumor reduction surgery, via the internal conjunctival route, given the internal extraconal location of the tumor.
Conclusion
NF2 is a severe disorder with a reserved functional prognosis. The main therapeutic goal is to maintain quality of life. The general principle is to treat only symptomatic or rapidly growing lesions. In all cases, clinical and radiological follow-up at least annually must be offered.
Conflicts of Interest
None.
Funding
None.
Article Info
Article Type
Case ReportPublication history
Received: Fri 17, Jul 2020Accepted: Tue 04, Aug 2020
Published: Mon 24, Aug 2020
Copyright
© 2023 Taoufik Abdellaoui. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited. Hosting by Science Repository.DOI: 10.31487/j.AJSCR.2020.03.11
Figures & Tables
Table 1: Manchester criteria for clinical diagnosis of NF2.
|
|
Additional findings needed for diagnosis |
|
Bilateral vestibular schwannomas |
None |
|
1st degree family relative with NF2 |
Unilateral vestibular schwannoma or two NF2-associated lesions (meningioma, glioma, neurofibroma, schwannoma, or cataract) |
|
Unilateral vestibular schwannoma |
Two NF2-associated lesions associated with the disorder (meningioma, glioma, neurofibroma, schwannoma, or cataract) |
|
Multiple meningiomas |
Unilateral vestibular schwannoma or two other NF2-associated lesions (glioma, neurofibromas, schwannoma, or cataract) |
References
- Evans DG, Huson SM, Donnai D, Neary W, Blair V et al. (1992) A genetic study of type 2 neurofibromatosis in the United Kingdom. I. Prevalence, mutation rate, fitness, and confirmation of maternal transmission effect on severity. J Med Genet 29: 841-846. [Crossref]
- Rouleau GA, Merel P, Lutchman M, Sanson M, Zucman J et al. (1993) Alteration in a new gene encoding a putative membrane-organizing protein causes neuro-fibromatosis type 2. Nature 363: 515-521. [Crossref]
- Lee JY, Kim H, Ryu CH, Kim JY, Choi BH et al. (2004) Merlin, a tumor suppressor, interacts with transactivation-responsive RNA-binding protein and inhibits its oncogenic activity. J Biol Chem 279: 30265-30273. [Crossref]
- Evans DG, Ramsden RT, Shenton A, Gokhale C, Bowers NL et al. (2007) Mosaicism in neurofibromatosis type 2: an update of risk based on uni/bilaterality of vestibular schwannoma at presentation and sensitive mutation analysis including multiple ligation-dependent probe amplification. J Med Genet 44: 424-428. [Crossref]
- Kluwe L, Mautner V, Heinrich B, Dezube R, Jacoby LB et al. (2003) Molecular study of frequency of mosaicism in neurofibromatosis 2 patients with bilateral vestibular schwannomas. J Med Genet 40: 109-114. [Crossref]
- Nunes F, MacCollin M (2003) Neurofibromatosis 2 in the pediatric population. J Child Neurol 18: 718-724. [Crossref]
- Evans DG, Huson SM, Donnai D, Neary W, Blair V et al. (1992) A clinical study of type 2 neurofibromatosis. Q J Med 84: 603-618. [Crossref]
- Bosch MM, Boltshauser E, Harpes P, Landau K (2006) Ophthalmologic findings and long-term course in patients with neurofibromatosis type 2. Am J Ophthalmol 141: 1068-1077. [Crossref]
- Ragge NK, Baser ME, Riccardi VM, Falk RE (1997) The ocular presentation of neurofibromatosis 2. Eye (Lond) 11: 12-18. [Crossref]
- Bouzas EA, Parry DM, Eldridge R, Kaiser Kupfer MI (1993) Visual impairment in patients with neurofibromatosis 2. Neurology 43: 622-623. [Crossref]
- Feucht M, Griffiths B, Niemuller I, Haase W, Richard G et al. (2008) Neurofibromatosis 2 leads to higher incidence of strabismological and neuro-ophthalmological disorders. Acta Ophthalmol 86: 882-886. [Crossref]
- Evans DG, Baser ME, O'Reilly B, Rowe J, Gleeson M et al. (2005) Management of the patient and family with neurofibromatosis 2: a consensus conference statement. Br J Neurosurg 19: 5-12. [Crossref]
- Lafford GH, Eccles SJ, Haq J, Orban N (2017) Sight preserving orbital decompression: a novel multidisciplinary approach to managing severe proptosis in neurofibromatosis type 2. BMJ Case Rep 2017: bcr2017221462. [Crossref]
- Lekovic GP, Schwartz MS, Hanna G, Go J (2018) Intra-Orbital Meningioma Causing Loss of Vision in Neurofibromatosis Type 2: Case Series and Management Considerations. Front Surg 5: 60. [Crossref]