Inguinal Dermatofibrosarcoma Protuberans: Clinical Case Presentation
A B S T R A C T
Dermatofibrosarcoma protuberans (DFSP) is a malignant skin neoplasm of slow growth characteristics. A case of inguinal DFSP is presented in order to review clinical diagnosis challenges, histological variants, malignancy behaviour and therapeutic modalities. Although rare in frequency of presentation, its high recurrence rate and metastatic potential make it an important tumor for the general surgeon to keep in mind.
Keywords
Dermatofibrosarcoma protuberans, inguinal tumors, skin tumors
Introduction
Dermatofibrosarcoma protuberans (DFSP) is a malignant skin neoplasm which can be easily misdiagnosed due to its rare occurrence and slow growth characteristics. While having a low malignancy grade, it has been reported to have a high local recurrence rate and some degree of metastatic potential. Therapeutic approach to such tumors may include wide excision as well as adjuvant radio or chemotherapy but the ample variety of presentations merits individualized care for each patient. The following case illustrates the most relevant aspects of DFSPs clinical and histopathological presentations and reviews its present therapeutic recommendations.
Clinical Case
A 45-year-old male presented with a focal, 3 cm, erythematous growth in the left inguinal region. The lesion had a nodular aspect, soft consistency, appeared to involve deeper tissues and was tender to touch (Figure 1). History revealed approximately 1 year of slow and progressive growth without other accompanying signs or symptoms. Preoperative diagnosis was unclear and decision was made to resect and analyse for further characterization. Wide excision was performed under local anaesthesia, obtaining a firm, well circumscribed mass of approximately 3x4 cm (Figure 2).

Figure 1: Left inguinal mass.
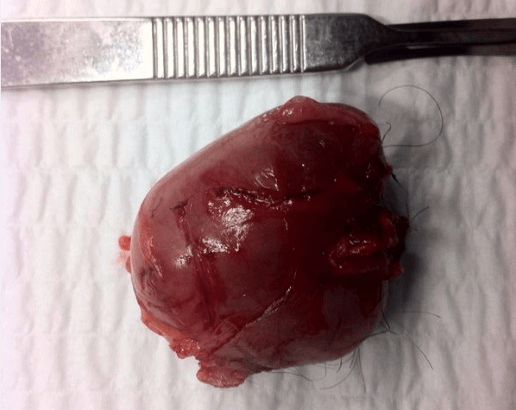
Figure 2: Complete tumor excision with overlying skin and no evidence of protuberances, tissue infiltration or satellite lesions.
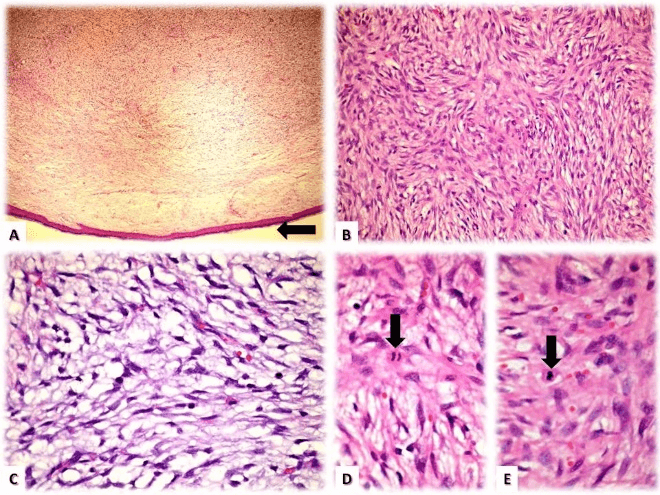
Figure 3: Microscopic analysis (Hematoxylin & Eosin stains): A) Sub-epidermic proliferation (arrow) of fusiform cells with well-defined margins. B) Classic ‘whirlpool’ cell distribution. C) Areas with low cellularity and abundant myxoid stroma. D & E) Isolated mitotic figures (arrows).
Pathologic analysis revealed a fibrohistiocytic dermal proliferation with cellular atypia and a moderate mitotic index (2-3 mitosis per 10 40x fields). The stroma had myxoid characteristics and ill-defined limits (Figure 3). Immunophenotype was defined by the following markers (Table 1 & Figure 4).
Table 1: Results from immunohistochemical analysis.
|
Positive markers |
Negative markers |
|
CD34, CD56, Ki-67 (10%) |
Protein S-100, Protein p53, CK AE1/AE3 |
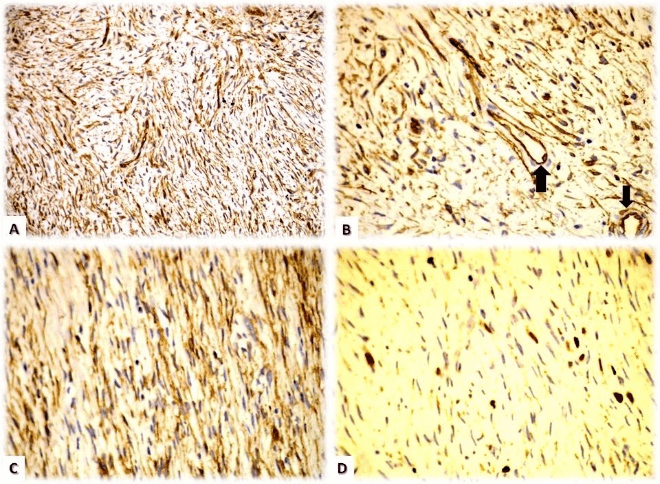
Figure 4: Immunohistochemical analysis: A) Intense and diffuse expression with CD34 on proliferating cells. B) CD34 positivity with internal control of positive endothelial (vascular) cells (arrows). C) CD56 expression on tumoral cells. D) Cellular proliferation index measured with Ki67 shows positivity in less than 5% of the tumoral cell nuclei.
The intense expression of CD34 and CD56, as well as the negativity of the remaining markers led to the diagnosis of a myxoid type dermatofibrosarcoma protuberans. After thorough explanation to the patient about recurrence and metastatic potentials, it was agreed to continue periodic follow-up without further treatment unless required. The patient has been free of disease for more than 3 years now.
Discussion
Initially described by Sherwell and Taylor in 1890 as a keloid sarcoma, DFSP has been coined several clinical names and may be found through different terms in the literature. In 1924 it was described as a recurrent fibrosarcoma of the skin and referred to as the Darier-Ferrand tumor after its describing authors. One year later, Hoffman was able to identify clinical and histologic differences between this type of tumor and other mesenchymal masses and introduced the term dermatofibrosarcoma protuberans. It is presently considered a sarcoma (mesenchymal origin neoplasia), that arises from the dermis, contains fibroblasts and histiocytes and typically presents with ramifications or protuberances which give it it’s infiltrative behaviour. Other terms that can be found to describe DFSP are malignant fibrous histiocytoma, hypertrophic morphea, fibroxanthoma and xanthogranuloma [1].
The origin of DFSP is unknown but most authors suggest that it arises from the transformation of fibroblasts into histiocytes. Other theories point to a neuroendocrine or undifferentiated mesenchymal cell origin. While some have hypothesized that DFSP´s tumoral growth may be triggered by small traumatic lesions, insect bites, skin burns, surgical scars or application of subdermal vaccines, most cases don´t have a history of such events [1]. On the other hand, the involvement of platelet derived growth factor (PDGF) has recently been implied in its pathogenesis. Cytogenetic studies have found that most cases of DFSP share a chromosomal translocation in chromosomes 17 and 22 that leads to the fusion of genes COL1a1 and PDGFb. These genes are responsible for the synthesis of type 1a1 collagen and the b chain of platelet derived growth factor, respectively. It is also believed that autocrine or paracrine stimuli that act upon the beta receptor of PDGF are what may unlock tumor growth and proliferation [2, 3].
Dermatofibrosarcoma protuberans may affect any race but is slightly more common in Afro-Americans. It is also more frequent in men than women (5:4 ratio) and usually affects young adults between the ages of 20 and 50 years. It represents only 0.1% of all malignant tumors and it is estimated to occur in 0.8 to 5 cases per million people [4]. Clinically it is characterized by being a slow growing tumor that appears subcutaneously. In 50 to 60% of cases it is located on the trunk or proximal extremities, 20 to 30% on the head and neck and 10 to 15% on the back. It appears as a solitary, multilobulated and well-defined tumor with soft surface and variable size. It may show white, rose, red or blue colouration.
Initially, DFSP may have an asymptomatic phase which can last several months or years, where the only sign is the presence of a hardened or sclerotic skin lesion. This may be followed by a more rapid growth phase where nodules, ramifications or protuberances can appear in the form of satellite lesions or deeper tissue involvement (including fascia, muscle or bone). Tumors may increase in diameter and present hyperesthesia, pain, ulceration or bleeding. Depending on the site, they may also provoke functional alterations, movement limitation and aesthetic deformity [1].
The formation of protuberances is thought to contribute to the infiltrative behaviour and to its locally malignant character of progressive growth and high recurrence rates which range from 20 to 50%, respectively. Factors related to risk of recurrence include grade of cellular pleomorfism and atypia, presence of tumor necrosis, a high mitotic rate and an incomplete surgical resection. Only 0.3% of cases have been reported to present lymphatic or hematogenous spread, and these have been associated with a rare form of fibrosarcomatous transformation seen in high grade sarcomas. Reports from the National Comprehensive Cancer Network (NCCN) show local recurrence rates that range from 0 to 60%, with regional and distant dissemination in 1 and 5%, respectively. Tumor staging for DFSP is classified as I (local disease), II (regional disease) and III (distant disease) [5]. Clinical diagnosis of DFSP may be challenging due to its rare occurrence. It is often misdiagnosed with other types of skin lesions such as dermoid cysts, neurofibromas, hydradenomas and keloid scarring. In its early phases it must be differentiated from scleroderma and in its more advanced forms, from Kaposi’s Sarcoma, lymphoma, leiomyosarcoma and basocellular carcinoma.
In its classical microscopic description, DFSP presents as a dermal proliferation with adipose tissue infiltration. It is formed by slightly pleomorphic fusiform cells arranged in a ‘whirlpool’ fashion. As shown in our example, they usually present isolated mitotic figures that do not exceed 5 in 10 40x fields and have no atypia. The stroma is usually formed by scarce collagenous tissue with mononuclear inflammatory cells and no associated necrosis. Variations from this classical presentation include a pigmented form, a myoid nodular form, a giant fibroblastoma form and myxoid stroma form. Finally, DFSP may present as a high-grade sarcoma (fibrosarcomatous DFSP) which has the worst metastatic potential [6].
Immunohistochemical studies are essential for precise diagnosis. The diffuse expression of CD34 antibodies on proliferating cells is characteristic (although this is an unspecific marker of endothelial cells) as well as the negativity of other antibodies such as S-100 protein (marker for melanomas and neural tumors), smooth muscle actin and factor VIII (vascular tumor markers). Other antibodies that may show positivity are EMA and CD56 but these tend to be more focal and variable. Figures 2A- 2C, illustrate the results in our case. From a histopathological stand-point, DFSP must be differentiated from other mesenchymatous lesions such as the cutaneous fibrohistiocytoma (dermatofibroma), the perineuroma and to a lesser extent, tumors from the peripheral nerve sheaths and smooth muscle [6].
Treatment of choice is centered on wide surgical excision. Unfortunately, because most preoperative diagnoses are unclear and satellite lesions or protuberances may be missed, initial excision is commonly incomplete. Large lesions usually require 3-5 cm margins, including the deeper tissues involved [5]. Fascia and muscle tissue may require en-bloc resection. When preoperative diagnosis is anticipated, a Mohs micrographic procedure may be the treatment of choice [4]. This technique, although highly complex, has shown recurrence rates as low as 2.4% (compared to 60% with incomplete resection and 23% with the typical wide resection). Lymphadenectomy is not usually required due to the low risk of dissemination through this route.
Other treatment modalities for DFSP have focused on adjuvant therapies. Traditionally, when a DFSP recurs or when wide resection is difficult due to tumor location (functional or aesthetic limitations), radiotherapy can been used. Approximately 5,000 to 6,000 cGy, fractioned into 200 cGy daily sessions, are applied on a 3-5 cm margin over the lesion or surgical scar. Unfortunately, radiotherapy has not shown significant tumor growth reduction and is not commonly used anymore [4]. On the other hand, knowledge of the association between DFSP and PDGFb receptor has led to more direct therapies. Imatinib (a tyrosine kinase inhibitor) and Sorafenib (an endothelial growth factor inhibitor) have shown promising results. Imatinib has been approved by the FDA for the treatment of some types of leukemia, gastrointestinal stromal tumors and now also for DFSP. It inhibits the receptor for PDGF and has shown tumor growth reduction in in vitro studies. It has also shown favourable results in recurrent or unresectable cases. Some tumors however may be resistant to treatment with Imatinib, especially those lacking the chromosomal translocation t(17;22) and therefore, some authors have suggested performing cytogenetic studies before initiating treatment [4]. Sorafenib, which has successfully been used in some types of angiosarcoma and neural origin sarcomas, has been reported to reduce tumor size in a patient with an Imatinib-resistant DFSP [7]. Further studies are required to confirm the benefit of these therapies in recurrent or unresectable DFSP cases.
Approximately 50 to 75% of local DFSP recurrences present within the first 3 years of resection and during this time, patients should be followed-up closely. Although recurrence after 10 years is very rare, at least 3 cases have been reported to do so at 13, 20 and 26 years, respectively [8]. Lymphatic metastasis occurs in 1% and hematologic dissemination (lung, brain and bone) in 3% of cases. Five-year survival has been reported at 99.2% but metastatic cases have a high mortality at two years [8]. Based on these findings, the NCCN recommends surgical site revision at 6 and 12 months with repeat biopsy in case of any suspicious lesion reappearance [5]. The case presented here has had no evidence of recurrence in more than 3 years follow-up. Based on the fact that the tumor was relatively small and completely excised, decision was consented with the patient not to undergo further treatment. Having widened the surgical site might have compromised the inguinal or genital areas and so was discarded. Use of radiotherapy or immunotherapy were also considered unnecessary because the histologic type (myxoid DFSP) has shown to have a very low probability of metastasis.
As is exemplified by this case, dermatofibrosarcoma protuberans represents a diagnostic challenge and surgeons must have a high index of suspicion in order to make a correct preoperative approach. Treatment modalities must be individualized depending on the site and size of the tumor, its infiltrative behaviour or the presence of satellite lesions, its histologic and immunologic pattern, and the appearance of recurrent or metastatic lesions. Close communication and patient follow-up are essential for good long-term results.
Article Info
Article Type
Case ReportPublication history
Received: Thu 17, Dec 2020Accepted: Fri 22, Jan 2021
Published: Sat 30, Jan 2021
Copyright
© 2023 Denzil Garteiz Martínez. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited. Hosting by Science Repository.DOI: 10.31487/j.SCR.2021.01.20
Figures & Tables
Table 1: Results from immunohistochemical analysis.
|
Positive markers |
Negative markers |
|
CD34, CD56, Ki-67 (10%) |
Protein S-100, Protein p53, CK AE1/AE3 |
References
- Rodríguez AM, González GM, Ramos GA (2003) Dermatofibrosarcoma protuberans. Rev Cent Dermatol Pascua 12: 2.
- Sirvent N, Maire G, Pedeutour F (2003) Genetics of dermatofibrosarcoma protuberans family of tumors: from ring chromosomes to tyrosine kinase inhibitor treatment. Genes Chromosomes Cancer 37: 1-19. [Crossref]
- Sjoblom T, Shimizu A, O’Brien KP, Pietras K, Dal Cin P et al. (2001) Growth inhibition of dermatofibrosarcoma protuberans tumors by the platelet derived growth factor receptor antagonist STI571 through induction of apoptosis. Cancer Res 61: 5778-5783. [Crossref]
- Mehrany K, Swanson NA, Heinrich MC, Weenig RH, Lee KK et al. (2006) Dermatofibrosarcoma protuberans: a partial response to imatinib therapy. Dermatol Surg 32: 456-459. [Crossref]
- Miller SJ, Alam M, Andersen JS, Berg D, Bickakjian CK et al. (2012) Dermatofibrosarcoma Protuberans. J Natl Compr Cancer Netw 10: 312-318. [Crossref]
- Serra Guillén C, Llombart B, Sanmartin O (2012) Dermatofibrosarcoma protuberans. Actas Dermosifiliogr 103: 762-777. [Crossref]
- Kamar FG, Kairouz VF, Sabri AN (2013) Dermatofibrosarcoma protuberans (DFSP) successfully treated with sorafenib: case report. Clin Sarcoma Res 3: 5. [Crossref]
- Lemm D, Mügge LO, Mentzel T, Höffken K (2009) Current treatment options in dermatofibrosarcoma protuberans. J Cancer Res Clin Oncol 135: 653-665. [Crossref]