Exploring the Dynamics of Nucleophilic Substitution Reactions: Understanding the Role of Entropy and Potential Energy in SN1 and SN2 Pathways
A B S T R A C T
The fundamental interplay between potential energy and entropy is a key facet of chemical reactions. Potential energy signifies stored energy in a chemical system, while entropy quantifies disorder. Understanding this connection is pivotal for comprehending reaction dynamics. This manuscript delves into the intricate relationship between potential energy and entropy, investigating their influence on chemical reactions. Potential energy arises from atomic and molecular positions, while entropy reflects molecular arrangement randomness. The equilibrium of these factors impacts reaction feasibility and kinetics. Through analysis, we explore how nucleophilic substitution reactions, essential for organic synthesis, showcase the potential energy-entropy interplay. Excessive potential energy with low entropy and excessive entropy both impede reactions. Favorable alignment of potential energy and entropy promotes reactions, yielding diverse energy states. Understanding potential energy and entropy interaction provides insights into reaction feasibility, rate, and control. This enriches foundational chemistry comprehension, facilitating efficient, predictable reaction design.
Keywords
Nucleophilic substitution, SN1, SN2, potential energy, entropy, mechanism
Introduction
Nucleophilic substitution reactions play a central role in the field of organic chemistry, governing the intricate processes of molecular modification and assembly. At the core of these reactions are nucleophiles, molecules that generously make their electron-rich centers available to enable the formation of new chemical bonds [1]. This fundamental reaction controls the assembly of organic structures ranging from basic compounds to complex biomolecules essential for sustaining life. At the core of nucleophilic reactions lies the dynamic interaction between nucleophiles and electrophiles. Electrophiles, characterized by their electron-deficient nature, engage in a captivating partnership with nucleophiles. This interaction results in the formation of new covalent bonds and the reorganization of atoms or groups [2].
In the fields of synthetic chemistry and pharmaceuticals, a comprehensive understanding of nucleophilic substitution reactions is of prime importance. Unraveling the mechanisms underlying these reactions and recognizing the factors that influence their kinetics and regioselectivity allow chemists to manipulate rational drug design. This in turn enables the design and production of innovative compounds tailored to specific structural properties and functionalities [3].
Our investigation takes an in-depth look at the intricacies of nucleophilic substitution reactions, examining them closely through a different perspective in particular from the view of entropy and potential energy, with an emphasis on clarifying the mechanisms underlying the SN1 and SN2 pathways [4]. Principles from thermodynamics and reaction energetics are interlaced together to elucidate the fundamental mechanisms, driving forces, and specific applications. By analysing these driving forces, we illuminate how nucleophilic reactions, which play a central role in organic chemistry, can be comprehensively understood by incorporating potential energy and entropy perspectives. Furthermore, we explore how modulation of these forces, whether at the level of the whole molecule or fraction components, can lead to a deeper understanding of the indispensable balance between these forces, a balance that fundamentally supports the precise control of reactivity [5-6].
Deciphering Potential Energy and Entropy in Reactions
In the broad field of chemistry, the delicate connection between two central concepts - potential energy and entropy - is at the core of the dynamics of chemical reactions. To comprehend and untangle this intricate connection, it's crucial to perceive potential energy as a reservoir of stored energy inherent to a chemical system. This energy is held within atoms and molecules due to their positions and interactions. From a molecular perspective, potential energy is embedded in the inherent constitution, intricate configuration, and unique conformations of molecules. The greater the degree of organization and rigidity of a compound, the higher its potential energy content [7-8]. Contrast this with entropy, a measure of randomness or disorder in a system. This concept of entropy is related to the flexibility, reorientation, and number of conformers that molecules possess. The more opportunities these molecules possess to disperse and rearrange themselves, the higher their entropy. The essence of entropy is also evident in the interplay of vibrations, rotations and translations within molecules [9-10].
Entropy represents molecular disorder and dissociation, while potential energy reflects organization and association. These factors are inversely related: higher potential energy corresponds to lower entropy and vice versa [11-12]. The duality of harmonic interplay between entropy and potential energy is essential and serves as a measure by which the intricate balance between molecular dispersion and organization within a given state can be remarkably quantified [13-14]. Potential energy stands as a key player in assessing a reaction's feasibility [15]. When potential energy is excessively high, a reaction may encounter difficulties progressing, when combined with low entropy. This situation signifies limited flexibility or a reduced availability of energy states for the reaction to occur [16]. On the other hand, excessively high entropy can also hinder reactions. If a molecule is excessively disordered and the potential energy is extremely low, the reaction might not proceed smoothly due to the molecule's high disorder and stability. However, when potential energy aligns favorably, reactions are more likely to unfold, particularly when accompanied by sufficient entropy or flexibility, leading to a variety of energy states [17].
Conversely, higher entropy signifies that molecules are more dispersed and less orderly, while lower entropy points to a more rigid arrangement. This molecular positioning profoundly affects the ease and speed of a reaction, consequently molecules with bulky substituents often possess higher potential energy due to limited flexibility and higher complexity, the contrary is true as well [18]. Harmonizing the interplay between potential energy and entropy is a key aspect of anticipating and enhancing chemical reactions. When these two factors work in synergy, reactions tend to unfold smoothly. This delicate balance affects both the overall favorability and kinetics of reactions and includes the interplay of molecular order and disorder [19]. The complicated interplay between potential energy and entropy is central to understanding and predicting chemical reactions. By decoding their interaction, chemists gain insight into the feasibility and rate of reactions. This deep understanding not only reveals the fundamental principles of chemistry, but also enables us to design reactions that are efficient, predictable, and precisely controlled.
Probing SN1 Reactions
Through the exploration of SN1 reactions, we acquire valuable insight into the characteristics of the starting materials. This understanding is closely related to reactivity, which depends on the complexity of the alkyl substituents neighbouring the carbon attached to the leaving group. Reactivity decreases in the order of tertiary, secondary and primary substrates. SN1 reactions use substrates characterized by a higher degree of complexity. This complexity results from the presence of carbon atoms in SN1 substrates, which exhibit an inherent inflexibility [20]. This rigidity results from the complicated three-dimensional arrangement of the carbon atom when it is bonded to the leaving group. Consequently, this structural constraint results in limited flexibility, leading to an increase in potential energy, a characteristic that adds a challenging energetic aspect to the reaction [21].
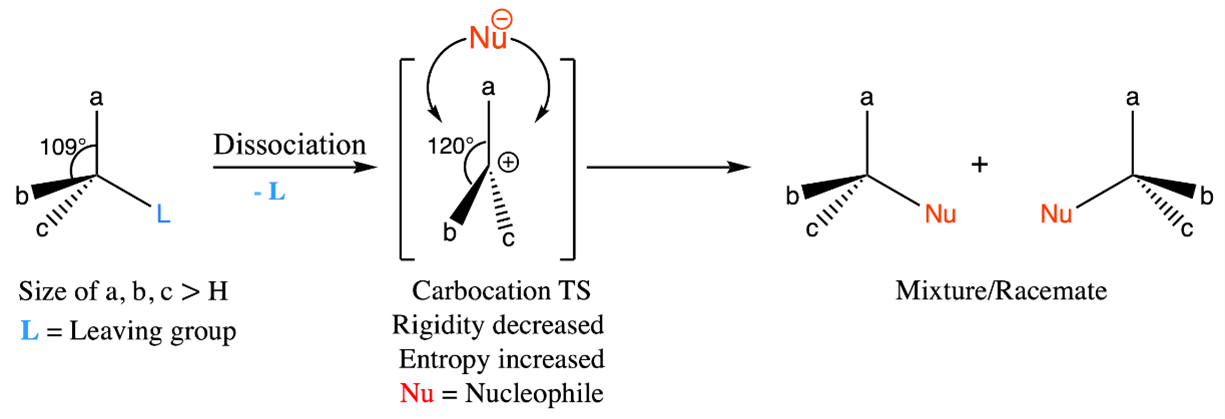
Figure 1: Adjustments in potential energy and entropy during SN1 reaction: transition state leading to induced flexibility and molecular freedom.
It is important to recognize the challenges that molecules with increased potential energy and decreased entropy pose to the progression of the reaction [22]. To overcome this hurdle, a strategic pathway is revealed during the formation of the transition state. This particular paradigm involves a core transformation in which a specific fragment (leaving group) of the molecule is dissociated (Figure 1). The rate determining step is the complete dissociation of the leaving group in the transition state. As a result, a carbocation is formed that is characterized by its planar geometry. This arrangement results in an expanded spatial domain around the carbocation, increasing the degree angle from 109° to 120°. This expansion enhances flexibility and reduces rigidity. This increased flexibility and entropy, while retaining a significant amount of potential energy, serves as a catalyst for the reaction [23-24].
As a result, a harmonic balance is reached, characterized by an increased entropy level and the retention of a considerable potential energy [25]. This state of balance proves to be a crucial driving force for the SN1 reaction to proceed. The driving force behind this reaction can be attributed to the increase in entropy that occurs during the transition state. This phenomenon leads to an acceleration in the reaction rate when higher temperatures are present, effectively promoting the dissociation process. However, as the entropy in the transition state increases, the selectivity of the reaction decreases. This result is due to the proliferation of different molecular orientations. This complicated interaction often results in the formation of a mixture or racemate, especially when the starting material exhibits chirality.
Probing SN2 Reactions
As we delve into the complicated nature of SN2 reactions, one important factor is the inherent simplicity of the starting material. In general, the substrate has a simple structure in which a carbon atom is closely linked to the leaving group. This structural aspect is critical for understanding reactivity patterns where primary carbons are more favourable for SN2 reactions compared to secondary carbons, while tertiary carbons exhibit a lower tendency for SN2 contribution [26-27].
At the core of this reactivity lies in the unique nature of the carbon attached to the leaving group, the point at which nucleophilic substitution occurs. This carbon, which is usually surrounded by smaller substituents such as hydrogen atoms, acquires a remarkable flexibility [20, 28]. This flexibility leads to higher entropy and relatively low potential energy. However, the conversion of a highly entropic molecule into a reactive moiety requires management of the potential energy enhancement. This precise management occurs during the formation of the transition state (Figure 2). At this crucial moment, the high flexibility of the parent molecule is restricted as the nucleophile interacts with the central carbon to create a more complex transition state. In the transition state of an SN2 reaction, a trigonal bipyramidal intermediate forms [29]. Here, the nucleophile attacks the carbon atom while the leaving group separates. This transitional configuration features the central carbon encircled by five substituents, two of which share partial bonds. This highly ordered arrangement leads to a rigidity that counteracts the earlier flexibility and results in increased potential energy. Consequently, the transition state becomes enriched in potential energy and becomes particularly rigid [30].
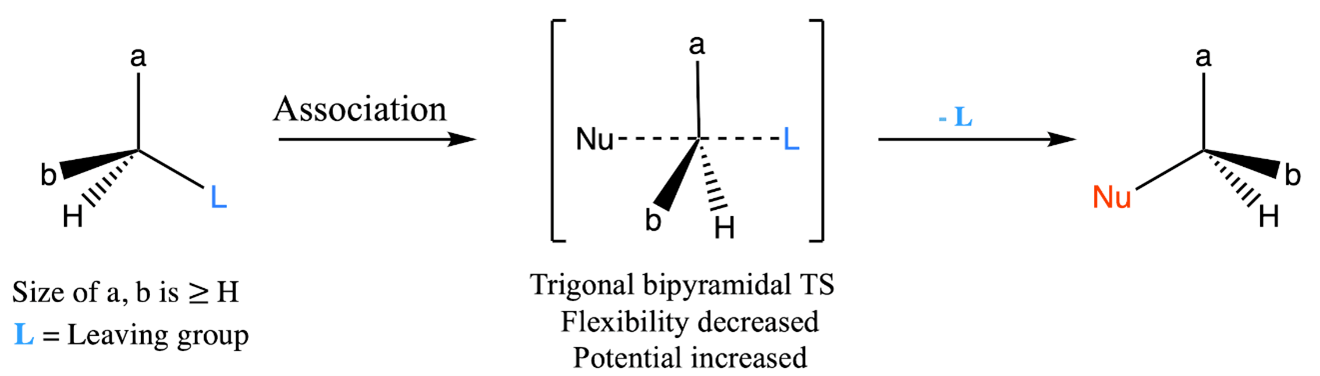
Figure 2: Adjustments in potential energy and entropy during SN2 reaction: transition state resulting in diminished flexibility and enhanced potential.
Due to the increased potential energy and rigidity, the reaction proceeds with improved selectivity. This selectivity often leads to the formation of a single product, often with an opposite configuration if the starting material was chiral. The essential assumption lies in the build-up of potential energy within the transition state, which directs the path of the reaction. However, it's essential to recognize that the formation of potential energy can be disrupted by factors that enhance flexibility. One notable factor is temperature, which plays a critical role. At higher temperatures, molecules have greater thermal energy, leading to more frequent molecular motion. This increased motion can lead to the destabilization of the transition state, as the geometry required for the trigonal bipyramidal arrangement becomes more difficult to achieve. As a result, the reaction might shift towards alternative mechanisms, such as E2 (elimination bimolecular) reactions. As a result, higher temperatures introduce movements that enhance flexibility, which could potentially disrupt the meticulously established potential energy configuration [31]. Therefore, carrying out SN2 reactions at room temperature or lower is more advantageous. In contrast to SN1 reactions, where elevated temperatures typically accelerate the reaction, the SN2 pathway deviates from this temperature-rate correlation. The intricate equilibrium between building potential energy and maintaining molecular rigidity outlines the unique reactivity of SN2 reactions.
Conclusion
In organic chemistry, nucleophilic substitution reactions play a crucial role in molecular transformation. This manuscript explores these reactions in relation to potential energy and entropy. Nucleophiles, electron-rich species, drive the formation of new bonds in compounds. The interplay of potential energy and entropy is examined, highlighting their reciprocal relationship. Excessive potential energy with low entropy or extreme entropy can hinder reactions. However, when potential energy aligns favorably with sufficient entropy, reactions thrive, resulting in diverse energy states. SN1 and SN2 reactions exemplify these principles. Transition states enable the overcoming of high potential energy and low entropy in SN1 reactions, while SN2 reactions benefit from simplicity and higher entropy. Understanding the interplay of potential energy and entropy enhances our comprehension of reaction feasibility, speed, and control. This knowledge empowers the design of efficient and predictable reactions, advancing the field of chemistry.
Acknowledgments
We would like to thank Prof. Quamrul Hasan, Prof. Khaled Abou Hadeed for their useful discussion and support.
Ethical Statement
The authors are accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
Conflicts of Interest
None.
Article Info
Article Type
Review ArticlePublication history
Received: Mon 21, Aug 2023Accepted: Wed 30, Aug 2023
Published: Mon 11, Sep 2023
Copyright
© 2023 Jawad Alzeer. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited. Hosting by Science Repository.DOI: 10.31487/j.AJMC.2023.01.02
Figures & Tables
References
1.
Pecher L, Laref S,
Raupach M, Tonner R (2017) Ethers on si(001): a prime example for the common
ground between surface science and molecular organic chemistry. Angew Chem
Int Ed Engl vol. 47: 15150-15154. [Crossref]
2.
Wang Y, Xu L, Szabó
I, Czakó G, Guo H et al (2016) Mode-specific SN2 reaction dynamics. J
Phys Chem Lett 17: 3322-3327. [Crossref]
3.
Şendur G (2021)
Representations in organic chemistry textbooks: nucleophilic substitution and
elimination reactions of alkyl halides. Turkiye Kimya Dernegi Dergisi Kısım
C: Kimya Egitimi 1: 71-92.
4. Florián J, Warshel A (1999) Calculations of hydration
entropies of hydrophobic, polar, and ionic solutes in the framework of the
langevin dipoles solvation model. J Phys Chem B 46: 10282-10288.
5.
Bento
AP, Bickelhaupt FM (2007) Nucleophilic
substitution at silicon (SN2@si) via a central reaction barrier. J
Org Chem 6: 2201-2207. [Crossref]
6.
Zhang
L, Zhang W, Liu J, Hu J (2009) C−f Bond cleavage
by intramolecular SN2 reaction of alkyl fluorides with o- and
n-nucleophiles. J Org Chem 7: 2850-2853. [Crossref]
7.
Becker NA, Cooper
MM (2014) College chemistry students' understanding of potential energy in the
context of atomic-molecular interactions. J Res Sci Teach 6: 789-808.
8.
Alzeer J (2022)
Directionality of chemical reaction and spontaneity of biological process in
the context of entropy. Int J Regenr Med 5: 1-7.
9.
McQuarrie DA, Simon
JD (1998) Physical chemistry: a molecular approach. Choice Reviews Online
5: 35-2708-35-2708.
10. Morán González L,
Besora M, Maseras,F (2021) Seeking the optimal
descriptor for SN2 reactions through statistical analysis of density
functional theory results. J Org Chem 87: 363-372. [Crossref]
11. Navamani K,
Rajkumar K (2022) Generalization on entropy-ruled
charge and energy transport for organic solids and biomolecular aggregates. ACS
Omega 31: 27102-27115. [Crossref]
12.
Yuan
S, Meijer BE, Cai G, Dixey RJC, Demmel F et al. (2022) Origin of the large entropy change in the molecular caloric and
ferroelectric ammonium sulfate. Adv Funct Materials 45: 2207717.
13.
Katsanikas
M, García Garrido VJ, Wiggins S (2020) The dynamical matching mechanism in phase space for caldera-type
potential energy surfaces. Chemical Physics Letters 137199.
14.
Alzeer J (2020)
Entropy and potential energy as a key role of Halalopathy in disease prevention
and cure. Longhua Chin Me 3: 20.
15.
Hebda JA, Aamold Z
(2019) Exploring biased probability using loaded dice: an active learning
exercise with analogy to entropic and energetic determinants of equilibria in
chemical systems. J Chem Educ 8: 1686-1690.
16. Bothe D, Dreyer W (2014) Continuum thermodynamics of
chemically reacting fluid mixtures. Acta Mech 6: 1757-1805.
17.
Xu
X, Zhou W, Jiang SP (2022) High‐entropy materials for water electrolysis. Energy Tech 11:
2200573.
18.
Xu
L, Yang L, Cao L, Li T, Chen S et al. (2013) Effect of bulky substituents on the self-assembly and mixing behavior of
arylene ethynylene macrocycles at the solid/liquid interface. Phys Chem Chem
Phys 28: 11748-11757. [Crossref]
19.
Alzeer J (2023) The
role of buffers in establishing a balance of homeostasis and maintaining
health. Am J Med Chem 4: 1-6.
20.
Ford GP, Scribner
JD (1990) Prediction of nucleoside-carcinogen reactivity. Alkylation of
adenine, cytosine, guanine, and thymine and their deoxynucleosides by
alkanediazonium ions. Chem Res Toxicol 3: 219-230. [Crossref]
21.
Alzeer J (2023)
Recognizing limitations: overcoming challenges in enhancing health and
preventing disease. Eur J Gen Med 3: 1-7.
22.
Alzeer J (2022)
Halalopathy: Improving potential energy and minimising entropy offer an
integrative approach for more effective treatment. Medicon Med Sci 2:
21-24.
23. Somalinga BR, Roy RP (2002) Volume exclusion effect as
a driving force for reverse proteolysis. Journal of Biological Chemistry
45: 43253-43261.
24.
Dehghani
MH, Pourhasan R, Mann RB (2011) Charged Lifshitz
black holes. Phys Rev D 84: 046002.
25.
Ge H, Qian H (2016)
Nonequilibrium thermodynamic formalism of nonlinear chemical reaction systems
with Waage–Guldberg’s law of mass action. Chemical Physics 241-248.
26. Fukuzumi S, Suenobu T, Hirasaka T, Arakawa aR, Kadish
KM (1998) Formation of c60 adducts with two different alkyl groups via
combination of electron transfer and SN2 reactions. J Am Chem Soc
36: 9220-9227.
27.
Liu
S, Hu H, Pedersen LG (2010) Steric, quantum,
and electrostatic effects on sn SN2 reaction barriers in gas phase. J
Phys Chem A 18: 5913-5918. [Crossref]
28.
Yu LJ, Coote ML
(2018) Electrostatic switching between SN1 and SN2
pathways. J Phys Chem A 2: 582-589. [Crossref]
29.
Zhang
M, Zhou M, Etten RLV, Stauffacher CV (1997) Crystal structure of bovine low molecular weight phosphotyrosyl
phosphatase complexed with the transition state analog vanadate. Biochemistry
1: 15-23. [Crossref]
30. Fang Y, Westaway KC (1991) Isotope effects in nucleophilic substitution reactions. viii. the effect of the form of the reacting nucleophile on the transition state structure of an SN2 reaction. Can J Chem 6: 1017-1021.
31. Chen Y, Cao Y, Shi Y, Xue Z, Mu T (2012) Quantitative research on the vaporization and decomposition of [emim][tf2n] by thermogravimetric analysis–mass spectrometry. Ind Eng Chem Res 21: 7418-7427.