Bilateral Wilms’ Tumor in a Female with Simpson-Golabi-Behmel Syndrome
A B S T R A C T
A 20-month-old female presented with a palpable abdominal mass. Imaging revealed bilateral renal masses and pulmonary nodules. The genetic evaluation identified a de novo 540kb X-chromosome deletion involving GPC3, PHF6, and HPRT1, and highly skewed X-inactivation (100:0). This clinical picture was suggestive of Wilms’ tumor in the setting of Simpson-Golabi-Behmel Syndrome.
Keywords
Simpson-Golabi-Behmel syndrome, Wilms’ tumor, X-linked recessive
Introduction
Simpson-Golabi-Behmel Syndrome (SGBS) is an X-linked recessive disorder caused by an alteration in the GPC3 or GPC4 gene [1]. The clinical picture is better understood in males due to the full penetrance of the disorder. Given generally milder clinical expressions of the disease, there is little information regarding expression in females, as is the case with many X-linked recessive disorders. Females are generally identified as carriers only after the diagnosis of an affected male relative [2]. Features of the disease in female carriers include tall stature and coarse facial features [1]. Males with full penetrance typically present with tall stature, coarse facial features, congenital heart defects, organomegaly, variable degrees of psychomotor impairment, and an increased risk of neoplasia such as embryonic tumors, primarily hepatoblastoma and Wilms’ tumors [1, 2].
There are a few explanations for full clinical expression of X-linked recessive conditions in heterozygous females. One such mechanism is skewed X inactivation in, which the wild-type gene is suppressed and a more severe phenotype results [2]. Other X-linked recessive single gene syndromes such as Börjeson-Fossman-Lehmann Syndrome (BFLS) and Lesch-Nyhan Syndrome (LNS) follow similar heredity patterns in females as SGBS and have all been associated with skewed (>80:20) or highly skewed (>90:10) X-inactivation ratios [3, 4].
Here we present a case of Wilms’ tumor with metastases to the lung in a 20-month-old female subsequently found to have a de novo deletion in the X-chromosome involving GPC3 (SGBS), PHF6 (BFLS), and HPRT1 (LNS) along with highly skewed X-inactivation (100:0). This patient represents the ninth instance of SGBS in females in the literature, the third to present with malignancy, and the second of which to have Wilms’ tumor. We also present a discussion of the implications of X-linked recessive disease identification in female patients.
Case Presentation
A 20-month-old female with developmental delay presented with a palpable abdominal mass. On computed tomography (CT), this was consistent with a large left renal mass (Figure 1) and was associated with smaller right renal masses, retroperitoneal lymphadenopathy, and pulmonary nodules. Biopsy of pulmonary nodules confirmed metastatic Wilms’ tumor without evidence of anaplasia. She received vincristine, doxorubicin, and dexamethasone (VAD) chemotherapy per AREN0534.
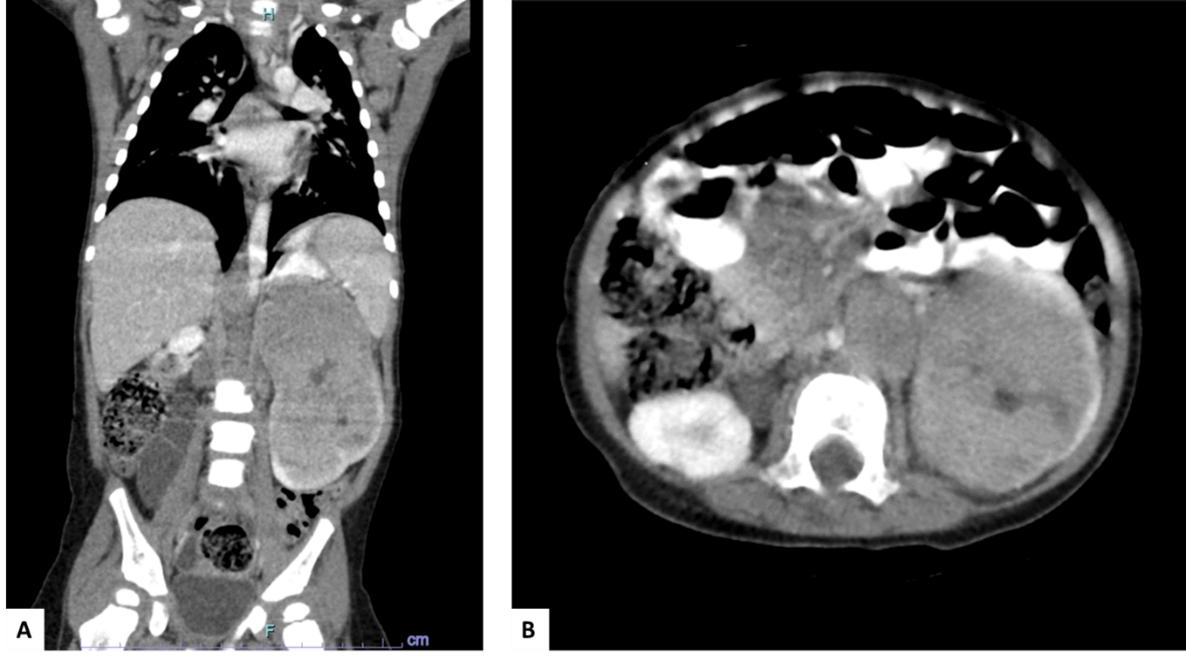
Figure 1: Computed tomography (CT) A) coronal and B) cross-section views pre-chemotherapy, illustrating a large solid left renal mass extending superiorly from the kidney and enlarged retroperitoneal lymph nodes.
Genetic evaluation of the patient identified an Xq26.2-q26.3 de novo 540 kb microdeletion of her X chromosome involving GPC3, PHF6, and HPRT1 as well as a highly skewed X-inactivation (100:0). She had no family history of chromosomal abnormalities or syndromes. Spinal imaging was negative for scoliosis. Echocardiogram revealed an atrial septal defect.
Post-chemotherapy imaging showed resolution of right renal masses. While the left renal mass decreased in size, there was evidence of extension to the hilum and collecting system. The patient thus underwent surgical management. Radical nephrectomy was performed as attempted nephron-sparing surgery could not preserve adequate renal vasculature for left kidney viability. The final pathology diagnosis was consistent with treated Wilms’ tumor with marked treatment effect and indicating only 5-10% viable tumor (Figure 2). The patient received left flank and pulmonary radiation as well as vincristine, dactinomycin, and doxorubicin (DD4A) chemotherapy. The patient is currently in remission and undergoing surveillance of right renal ultrasounds.
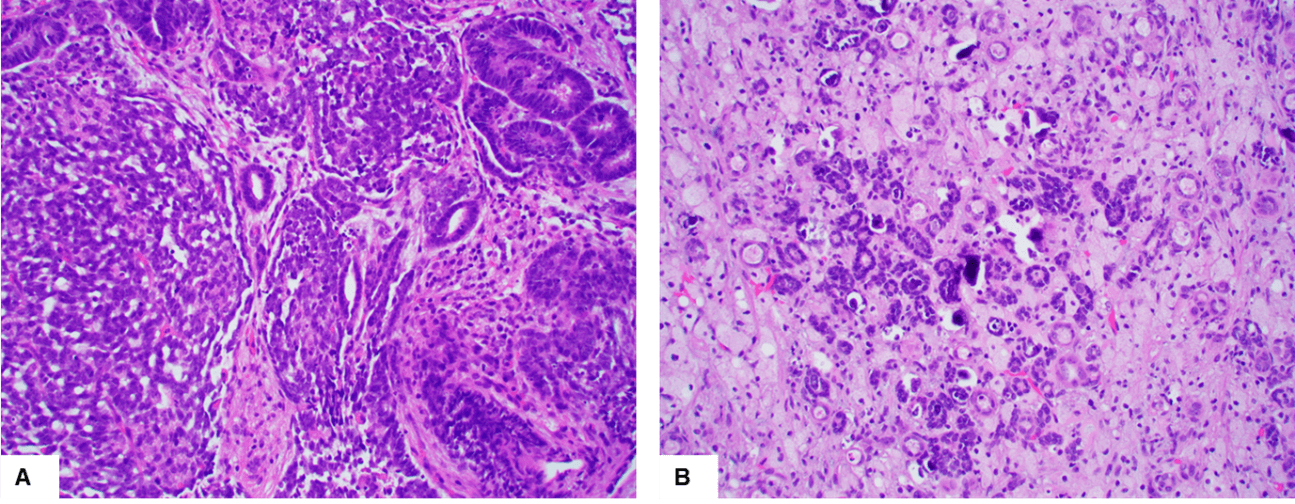
Figure 2: Representative H&E staining from A) lung nodule and B) treated left kidney mass. The lung nodule is composed of epithelial elements (manifest as well-formed tubular structures) and blastemal elements. The treated kidney mass shows aggregates of small nephroblastic tubules admixed with histiocytes and scattered calcification. Original magnification: 200 X for both A&B.
Discussion
SGBS is an X-linked disorder characterized by overgrowth, developmental delay, congenital heart defects, renal anomalies, facial dysmorphia, and embryonic tumors, specifically hepatoblastoma, and Wilms’ tumor [1]. Female carriers of SGBS typically have milder features, as they may not exhibit full penetrance [2]. Females with the full SGBS phenotype represent extremely rare exceptions to the X-linked recessive disorders model, with only eight previously published cases of symptomatic SGBS in females [1, 2]. The first such female case of SGBS was reported in 1994, involving mutations at chromosome Xq25-27 and a balanced X; 1 translocation [2, 5]. Unlike male patients, females with full penetrance of SGBS generally must demonstrate other alterations in the expression of the X chromosome. For example, four of the eight female patients with the full SGBS phenotype were also found to have X: autosome translocation, while no translocations were found in a study of approximately 250 affected males [2]. An alternative inheritance pattern involves gene duplication involving GPC3 and GPC4, the only two genes in which mutations are known to cause Simpson-Golabi-Behmel syndrome [1]. Duplication of either gene can result in a frameshift and subsequently expression of a premature termination codon, resulting in a truncated and, in turn, non-functional protein. Four out of eight symptomatic SGBS females showed duplications in either the GPC3 or GPC4 genes [1]. Finally, highly skewed X-inactivation can also be responsible for the presentation of SGBS females. This inheritance pattern is responsible for the more severe phenotype seen in the patient we describe above.
It is important to also note that of the eight cases described thus far, two were associated with malignancies, one being Wilms’ tumor [1]. This could imply that females who present with full penetrance of SGBS may be at high risk of developing the oncologic complications of SGBS. The two patients with the oncologic manifestations of SGBS who have been described in the literature also demonstrated duplication of SGBS1 [1]. The patient we describe, who presented with de novo microdeletion of the affected gene with severely skewed x-inactivation, may thus represent a novel inheritance pattern that is associated with oncologic manifestations in female patients with SGBS.
Conclusion
Here, we present a rare female variant of SGBS with bilateral Wilms’ tumor. Severely skewed X-inactivation is an unusual mechanism for the full expression of x-linked genetic disorders such as SGBS in females.
Funding
None.
Abbreviation
BFLS: Börjeson-Fossman-Lehmann Syndrome
CT: Computed Tomography
DD4A: Vincristine, Dactinomycin, and Doxorubicin
LNS: Lesch-Nyhan Syndrome
SGBS: Simpson-Golabi-Behmel Syndrome
VAD: Vincristine, Doxorubicin, and Dexamethasone
Article Info
Article Type
Case ReportPublication history
Received: Wed 06, Jul 2022Accepted: Thu 21, Jul 2022
Published: Thu 04, Aug 2022
Copyright
© 2023 Zoe Baker. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited. Hosting by Science Repository.DOI: 10.31487/j.SCR.2022.08.01
Figures & Tables
References
1. Schirwani S,
Novelli A, Digilio MC, Bourn D, Wilson V et al. (2019) Duplications of GPC3 and
GPC4 genes in symptomatic female carriers of Simpson-Golabi-Behmel syndrome
type 1. Eu J Med Genet 62: 243-247. [Crossref]
2. Vaisfeld A, Pomponi
MG, Pietrobono R, Tabolacci E, Neri G et al. (2017) Simpson-Golabi-Behmel
syndrome in a female: A case report and an unsolved issue. Am J Med Genet A
173: 285-288. [Crossref]
3. Gécz J, Turner G,
Nelson J, Partington M (2006) The Börjeson-Forssman-Lehman
syndrome (BFLS, MIM #301900). Eur
J Human Genet 14: 1233-1237. [Crossref]
4. Torres RJ, Puig JG (2007) Hypoxanthine-guanine phosophoribosyltransferase (HPRT) deficiency: Lesch-Nyhan syndrome. Orphanet J Rare Dis 2: 48. [Crossref]
5. Punnett HH (1994) Simpson-Golabi-Behmel syndrome (SGBS) in a female with an X-autosome translocation. Am J Med Genet 50: 391-393. [Crossref]