Journals
An exploration of interactions between the antiarrhythmic drug dronedarone and hERG potassium channel pore
A B S T R A C T
Dronedarone is a non-iodinated analogue of the Class III antiarrhythmic agent amiodarone. It exerts potent inhibition of “hERG” potassium channels that underpin the cardiac rapid delayed rectifier potassium current, IKr. This study aimed to extend understanding of interactions between dronedarone and the hERG channel. Whole-cell patch-clamp recordings were made at 37C of hERG channel current (IhERG) from HEK-293 cells expressing wild-type (WT) hERG or alanine mutants of residues in the channel’s pore-helix/selectivity filter region (T623, S624, V625) or S6 helices (S649, Y652, F656, V659). Molecular docking simulations were performed using a cryo-EM structure of hERG and a MthK-based homology model. The half-maximal inhibitory (IC50) value for WT IhERG inhibition by dronedarone was 42.6 ± 3.9 nM (n= at least 5 cells for each of 6 concentrations). 600 nM dronedarone exerted reduced WT IhERG block when the direction of K+ flux was reversed, consistent with interactions between the drug and permeant ion. In contrast with recently reported data for amiodarone, the S624A mutation did not attenuate IhERG blockade, whilst T623A and V625A channels exhibited modestly attenuated block. The S649A mutation was without significant effect and the Y652A and F656A mutations exhibited modest reductions in block. The V659A mutation produced the most marked effect on dronedarone action. Docking simulations were generally consistent with modest interactions with canonical binding residues and suggested an indirect rather than direct effect of the V659A mutation on the drug’s action. These findings leave open the possibility that as yet unexplored residue(s) could act as key determinants of high affinity hERG channel block by dronedarone.
K E Y W O R D S
Amiodarone, antiarrhythmic, Class III, Dronedarone, hERG, QT interval, potassium channel
I N T R O D U C T I O N
The cardiac rapid delayed rectifier potassium current, IKr is critical for normal ventricular repolarization [1]. The channels underlying IKr are encoded by hERG (human ether-à-go-go Related Gene; alternative nomenclature KCNH2) [2,3]. Loss-of-function hERG¬ mutations underlie the LQT2 form of congenital long QT syndrome, whilst gain-of-function mutations give rise to the SQT1 form of congenital short QT syndrome [4, 5]. IKr/hERG channels are also key drug targets in the heart: they represent the major potassium channel target for a number of antiarrhythmic drugs and are also sensitive to pharmacological inhibition by structurally and therapeutically diverse drugs that are associated with the drug-induced (acquired) form of long QT syndrome [4, 6, 7].
Amiodarone is one of the most effective antiarrhythmic drugs in clinical use. It can be used in patients with structural heart disease and be helpful in long term treatment of atrial fibrillation (AF) [8]. Intravenous amiodarone is valuable in the treatment of life-threatening ventricular arrhythmias [9]. Amiodarone exerts Class I-IV antiarrhythmic actions through effects on multiple targets [10, 11]. Unfortunately, amiodarone can produce a number of extra-cardiac side effects, some of which may result from the incorporation of iodine in its structure; dronedarone is a non-iodinated benzofuran analogue of amiodarone that was developed to retain antiarrhythmic effectiveness whilst reducing unwanted side effects [11, 12]. Dronedarone is an effective inhibitor of native cardiac IKr and in experiments on recombinant hERG and KCNQ1+KCNE1 channels (representing respectively IKr and IKs) dronedarone was observed to show selectivity for hERG over KCNQ1+KCNE1 [13-15]. When tested against hERG in a mammalian expression system, dronedarone inhibited hERG current (IhERG) with a half-maximal inhibitory concentration (IC50) below 100 nM, making it a high potency hERG channel inhibitor [16].
The archetypal high affinity Class III drug hERG inhibitors come from the methanesulphonanilide class (which includes dofetilide, MK499 and E-4031). Experiments with these agents have revealed key drug binding determinants on the S6 helices and pore-helix/close to the selectivity filter [17, 18]. Prominent amongst these are two S6 aromatic residues, Y652 and F656, mutation of which drastically reduces methanesulphonanilide binding [17, 18]. Indeed, for the majority of drugs examined in detail one or both of these aromatic residues have been found to be key binding determinant(s) [4, 7]. Experiments on dronedarone, however, showed that a “profound” IhERG blocking concentration (>65-fold the wild-type channel IC50) was able effectively to inhibit hERG channels in which Y652 or F656 had been mutated, raising questions regarding the dronedarone binding site on hERG [16]. Recently, we published a detailed analysis of binding to hERG of structurally related amiodarone, showing that amiodarone can bind within the hERG channel pore [19], but that the relative importance of particular binding residues differs from that previously reported for other high affinity agents [17-19]. To complement this, the purpose of the present study was to evaluate the impact on dronedarone’s inhibition of IhERG of a total of 7 mutations of pore-helix and S6 residues implicated in drug binding. Our results provide further evidence of variation in overall composition of the binding site(s) for different hERG blocking drugs.
Materials and Methods
Mutagenesis
The residues examined in this study are highlighted in Figure 1A. Alanine mutants of hERG at the base of the pore helices near the selectivity filter (T623A, S624A, V625A) and the S6 helix (S649A, V659A), were constructed using the QuickChange® site-directed mutagenesis kit (Stratagene, La Jolla, CA) as previously reported [19-21]. Mutations were confirmed by sequencing the entire open reading frame (Eurofins MWG Operon, Ebersberg, Germany).

Figure 1: (A) Sequence alignment for hERG and the MthK channel, highlighting the pore helix and S6 transmembrane domains. The residues of hERG analysed in this study by alanine mutagenesis are underlined. Bottom row of symbols highlights amino acid identities (-) and strong similarities (:). Amino acids in grey font have side chains facing the pore cavity of the MthK structure. Note that the last four residues of S6 in the MthK structure italicised (INRE) are not seen in the crystal structure and aren’t included in the hERG model.
(B) Representative traces of WT IhERG elicited by voltage protocol (lower panel, start to start interval between successive application was 12s) in control solution and after reaching steady-state, 10 minutes following exposure to 30nM Dronedarone (Droned).
(C) Concentration-response relationship for inhibition of WT IhERG by Droned, with fractional block values corrected for run-down as indicated in the Methods. The half-maximal inhibitory (IC50) value for IhERG inhibition by Droned was 42.6 ± 3.9 nM and Hill -coefficient was 0.93±0.07 (n= at least 5 cells for each of 6 concentrations tested).
(D) Bar chart comparing the mean fractional block of outward and inward WT IhERG tails in 4mM [K+] and in 94mM [K+] produced by 600nM Droned, *** p < 0.001, one-way ANOVA followed by Bonferroni’s post-test.
Maintenance of mammalian cell lines and cell transfection
HEK 293 cells (European Collection of Cell Cultures, Porton Down, UK) were maintained at 37 °C, 5% CO2 in Dulbecco's minimum essential medium with Glutamax-1 (DMEM; Gibco, Paisley, UK). This was supplemented with 10% foetal bovine serum (Gibco, Paisley, UK). Cells were transiently transfected with cDNA plasmids encoding wild-type (WT) and mutant hERG using Lipofectamine 2000 (Invitrogen, Paisley, UK) according to the manufacturer's instructions. Expression plasmid encoding CD8 was also added (in pIRES, donated by Dr I Baró, University of Nantes, France) to be used as a successful marker of transfection. Recordings were performed 24–48 h after transfection. Successfully transfected cells (positive to CD8) were identified using Dynabeads® (Invitrogen, Paisley, UK) [19- 22]. The HEK 293 cell lines stably expressing mutant F656A and Y652A hERG created in our laboratory have been previously reported [23]. Cells were passaged using enzyme free cell dissociation solution (Millipore, Watford, UK) and plated onto sterilised 13-mm glass coverslips in 40-mm petri dishes containing DMEM, which was supplemented with 10% foetal bovine serum, 100 µg/mL of hygromycin [19, 23].
Solutions, electrophysiological recordings, experimental protocol and data analysis
Measurements of hERG current (IhERG) were made at 37 ± 1 °C as described previously [19-21]. Cells were superfused in the recording chamber with normal Tyrode’s solution containing (in mM): 140 NaCl, 4 KCl, 2.5 CaCl2, 1 MgCl2, 10 Glucose, and 5 HEPES (titrated to pH of 7.45 with NaOH). Patch-pipettes (Corning 7052 glass, AM Systems, Carlsborg, USA) were pulled and heat-polished (Narishige MF83, Tokyo, Japan) to 2.5–4 MΩ; pipette dialysate contained (in mM): 130 KCl, 1 MgCl2, 5 EGTA, 5 MgATP, 10 HEPES (titrated to pH 7.2 using KOH) [19-21]. Dronedarone HCl (Sequoia Research Products Ltd, Pangbourne,UK) was dissolved in ethanol to produce a stock solution of 50 mM, which was serially diluted to produce stock solutions ranging from 50 mM to 5 μM. The stock solutions were then diluted 1:1000-fold with Tyrode’s solution to achieve concentrations stated in the results section.
Whole cell patch clamp recordings of membrane currents were made using an Axopatch 200A amplifier (Axon Instruments, Foster City, CA, USA) and a CV201 head stage. Between 70% and 80% of the electrode series resistance could be compensated. Data were recorded via a Digidata 1440A interface (Molecular Devices, Sunnyvale, CA, USA) and stored on the hard disk of a personal computer. Data digitization rates were 10 kHz during voltage protocols, and a bandwidth of 2 kHz was set on the amplifier.
As in previous similar structure function studies [19-21,24,25], activating voltage commands to +20 mV were used, with tail currents observed at either −40 mV, or −120 mV (for T623A, V625A, F656A, V659A). High external 94 mM [K+] (with NaCl concentration correspondingly reduced) conditions were used for comparatively poorly expressing mutations (T623A and F656A). For all mutants studied, block levels were attained by repetitive stimulation for 10 min and fractional inhibition of IhERG tails measured. The data for each mutant were compared with WT IhERG studied under comparable conditions [19-21, 24, 25]. Data were analysed using Clampfit 10.3 (Molecular Devices (UK) Limited, Wokingham, UK), Excel 2013 (Microsoft, Redmond, WA), Origin 7 (OriginLab Corporation, Northampton, MA, USA), and Graphpad Prism 7.0 (Graphpad Inc, La Jolla, CA, USA) software. The data are presented as the mean ± standard error of the mean (SEM). Statistical comparisons were made using a one-way analysis of variance (ANOVA) followed by a Bonferroni post-test, as appropriate. p values < 0.05 were taken as being statistically significant.
Concentration–response data and correction for IhERG run-down
The fractional block (FB) of IhERG “tails” by the different drug concentrations studied was determined using the equation:
Fractional block = 1 - ((IhERG-Droned)/IhERG-control) (1)
where “Fractional block” refers to the degree of inhibition of hERG current by a given concentration of Dronedarone. IhERG-Droned and IhERG-control represent “tail” current amplitudes in the presence and absence of dronedarone.
Concentration–response data were fitted by a standard Hill equation of the form:
Fractional block = 1/ (1 + (IC50/[Droned]) h) (2)
where IC50 is [Droned] producing half-maximal inhibition of the IhERG tail and h is the Hill coefficient for the fit.
Like amiodarone and its relatives, dronedarone is lipophilic and exhibits a progressive development of IhERG blockade, reaching a stable level of block by ∼10 min of drug exposure, with continuous application throughout this period of the voltage protocol [16, 19, 26]. During this period, there was some overlying rundown of IhERG. Therefore, to correct IhERG rundown we adopted a method used previously [19, 26], in which we subtracted 12.8% (representing the mean rundown recorded over 10 minutes in control experiments) of current magnitude from the last tail current in the control periods and used the resulting value to calculate fractional block following (10 min) exposure to dronedrone.
Computational docking simulations
Docking simulations were done using the recent open pore cryo-EM structure of a hERG construct (PDB: 5VA1) [27] and an open pore homology model of hERG constructed on the structure of MthK (PDB: 1LNQ) [28] as described previously [29-30]. Docking was performed using GOLD version 5.6 (Cambridge Crystallographic Data Centre, Cambridge, UK) with the ChemPLP scoring function as described previously [30].
Table 1: Summary data showing mean fractional block levels for WT and mutant hERG channels with 600 nM dronedarone
|
Channel |
Voltage step |
K+ |
numbers |
Fractional block |
p value |
|
|
mV |
mM |
|
Mean ±SE |
|
|
WT-1 |
-40 |
4 |
5 |
0.97±0.01 |
|
|
WT-2 |
-120 |
4 |
5 |
0.82±0.02 |
p<0.001 v WT-1 |
|
WT-3 |
-120 |
94 |
5 |
0.75±0.01 |
p<0.001 v WT-1 |
|
T623A |
-120 |
94 |
5 |
0.58±0.04 |
p<0.01 v WT-3 |
|
S624A |
-40 |
4 |
5 |
0.92±0.02 |
p>0.05 v WT-1 |
|
V625A |
-120 |
4 |
5 |
0.63±0.03 |
p<0.05 v WT-2 |
|
S649A |
-40 |
4 |
6 |
0.93±0.02 |
p>0.05 v WT-1 |
|
Y652A |
-40 |
4 |
6 |
0.76±0.06 |
p<0.005 v WT-1 |
|
F656A |
-120 |
94 |
5 |
0.46±0.04 |
p<0.001 v WT-3 |
|
V659A |
-120 |
4 |
5 |
0.36±0.07 |
p<0.0001 v WT-2 |
Results
WT IhERG inhibition by dronedarone
A standard voltage step protocol, comprised of a 2-second depolarization to + 20 mV followed by repolarization to − 40 mV was used to elicit IhERG tails [19, 21, 26]. Tail current amplitude was measured relative to current elicited by a brief (50 ms) pre-pulse prior to the + 20 mV test command [19, 21, 26]. Figure 1B shows example traces of IhERG in control and in the presence of 30nm dronedarone (giving 47% blockade of the IhERG tail for this cell, following run-down correction). A total of 6 dronedarone concentrations were tested (extending concentration-response information previously based on 5 concentrations) [22]. Figure 1C shows the mean concentration-response relation for WT IhERG (with fractional block at each concentration plotted against the Log [Dronedarone]). The data were fitted with equation 2 (Materials and Methods), yielding IC50 and Hill co-efficient values of 42.6 ± 3.9 nM and 0.9±0.07
We then performed experiments in which the direction and magnitude of K+ flux was altered. Figure 1D shows that 600 nM dronedarone produced a substantial fractional block of 0.97±0.01 (n=5) for outward IhERG tails in 4 mM [K+]e. Consistent with a previous report [16], when measuring inward IhERG tails at 120 mV in lower panel in Figure 2A, the extent of inward IhERG tail inhibition by 600 nM dronedarone was less extensive than that seen for the outward tail current, fractional block reduced to 0.82±0.02 (n=5), and reduced even further to 0.75±0.01 (n=5) for inward IhERG tails in raised 94 mM K+ respectively (both p < 0.001 compared with outward current in 4mM [K+], data summarised in Figure 1D and Table 1).
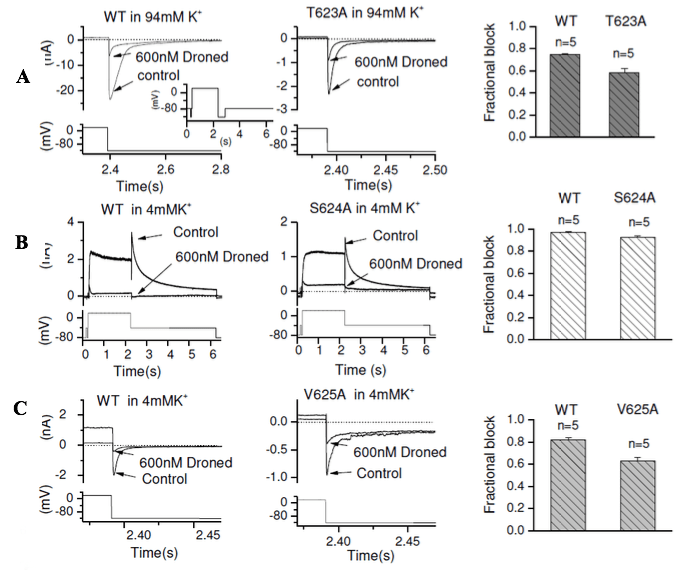
Figure 2: Representative IhERG traces from WT hERG (left panel of Figure 2A, B, C) and pore helix mutant T623A (middle panel of A), S624A (middle panel of B) and V625A (middle panel of C) in the absence and presence (following 10 min exposure) of 600nM of dronedarone (Droned) recorded under the same recording condition by using the same voltage protocol shown in each lower panel. Inward currents were elicited by the voltage protocol as shown as an inset to left panel of A. The expanded traces (and corresponding portion of the voltage protocol) are shown for clarity of display. The right panel of A, B and C show bar charts displaying the mean fractional block of mutant compared with its WT control. Statistical significance of comparisons of these values is given in Table 1.
Effects of pore helix mutations on IhERG inhibition by dronedarone
Amino acids located at the base of the pore helix (T623, S624 and V625) have been identified as important components of drug-binding sites for some hERG blockers [18,19,31-33]. To investigate the roles of these residues in dronedarone block of IhERG, the T623A, S624A and V625A mutations were studied. As previously [19], the T623A mutant was studied by measuring the inward IhERG tail current at −120 mV in raised (94mM) [K+]e. WT and T623A inward IhERG tails were recorded, with examples shown in Figure 2A (WT, left panel; T623A, middle panel) in the absence and presence of 600nM dronedarone, after 10 minutes exposure, with whole voltage protocol shown as left inset. The mean fractional block values are shown in the right panel of Figure 2A and are also given in Table 1, showing a modest but significant reduction in block for T623A IhERG. Our previous report showed that the pore helix S624A mutant produced substantial attenuation of the blocking effect of the structurally related hERG blocker amiodarone [19]. The effect of S624A hERG on IhERG block by dronedarone is shown in Figure 2B, in which block of WT IhERG (left panel) and S624A IhERG (middle panel) with the standard protocol were compared by recording outward hERG current in normal 4mM [K+] e. As shown in the representative traces and mean data (Figure 2B right panel), and summarised in Table 1, there was no significant effect of the S624A mutation on IhERG block by dronedarone. V625 is located within the K+ signature sequence of hERG (SVGFG) and mutation to alanine reduces the selectivity of hERG channel for K+. This mutant requires to be studied by measuring the inward hERG tail current at −120 mV (e.g. [19]). Figure 2C contains representative traces for WT (left panel) and V625A (middle panel) IhERG inhibition by dronedarone, with mean data shown in the bar graph in the right panel of Figure 2C and summarised in Table 1. The V625A mutation produced a modest, but significant reduction in dronedarone action.
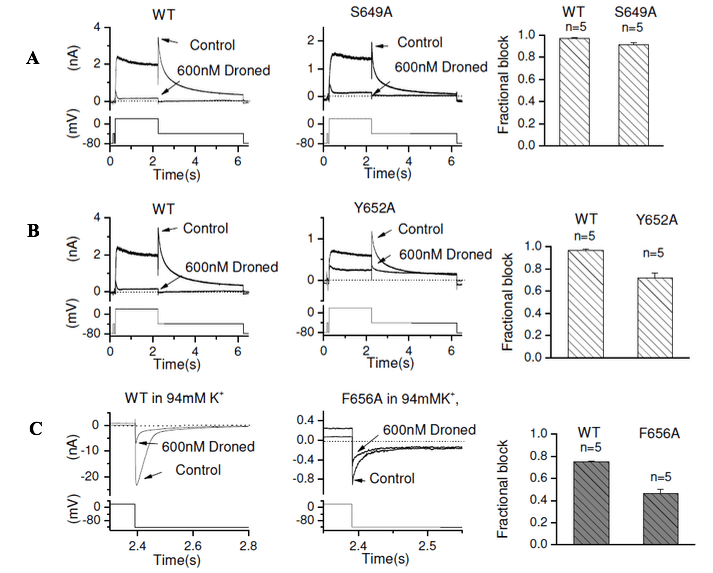
Figure 3: Representative IhERG traces from WT-hERG (left panel of A, B, C) and S6 mutants S649A (middle panel of A), Y652A (middle panel of B) and F656A (middle panel of C) in the absence and presence (following 10 min exposure) of 600nM of dronedarone (Droned) elicited by the voltage protocol shown as lower traces in each panel. The right panels of A, B and C show bar charts displaying the mean fractional block of mutant compared with its WT control. Statistical significance of comparisons of these values is given in Table 1.
IhERG inhibition of the S6 pore helix mutations by dronedarone
The interaction between dronedarone with each of the S649A and Y652A hERG mutants was examined using the standard protocol and recording conditions [19]. Figure 3A shows representative traces for WT (left panel) and S649A (middle panel) IhERG in the absence and presence of 600 nM dronedarone, with mean data summarised in the bar chart in Figure 3A right panel and Table 1. There was no significant reduction in IhERG block with the S649A mutation. For the Y652A mutation (shown in Figure 3B), there was a modest but significant reduction in the extent of IhERG block with dronedarone (see also Table 1).
The F656A mutant is associated with low levels of membrane-expression; IhERG measurements from F656A were facilitated by measuring inward tail currents elicited at 120 mV in the presence of higher external (94 mM) [K+] (using the same protocol as for T623A). Figure 3C shows the effects of 600nM dronedarone on WT and F656A inward IhERG tails, with mean data shown in the bar graph in the right panel and summarised in Table 1. There was a modest but significant reduction in block of IhERG by dronedarone for F656A hERG compared to its WT control.
Significant reduction of IhERG inhibition by the V659A mutation
The most marked reduction of IhERG inhibition was with the V659A S6 mutant. Figure 4Ai shows representative traces for WT and V659A IhERG in normal 4mM [K+] e solution in the absence and presence of 600nM dronedarone. Figure 4Ai shows traces with the entire voltage protocol, whilst Figure 4Aii shows expanded areas of the traces, focusing on the inward tail current, during which fractional block was measured. Figure 4B and Table 1 show mean data, indicating that the level of inhibition was considerably attenuated for this mutation.
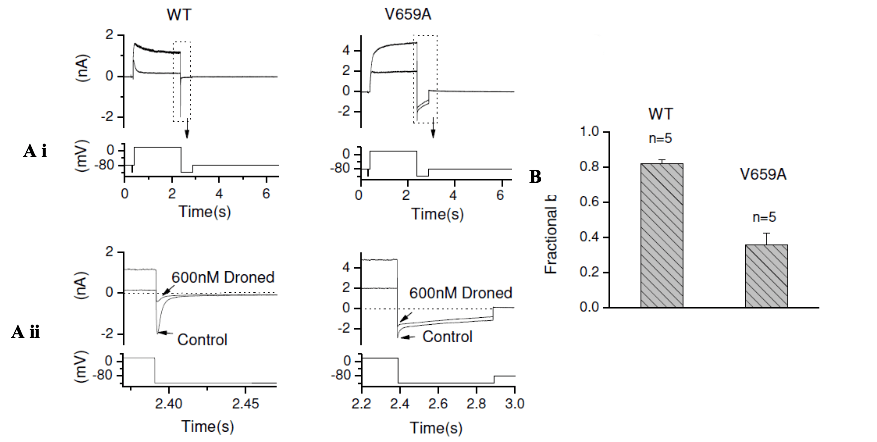
Figure 4: Representative WT hERG (left panel of Ai) and the S6 mutant V659A (right panel of Ai) in the absence and presence (following 10 min exposure) of 600nM of dronedarone (Droned) elicited by the voltage shown under the current traces. Aii shows expanded views of tail currents aligned with the corresponding portion of the voltage protocol (note different timescales for WT and V659A traces). (B) Bar charts displaying the mean fractional block of V659A IhERG compared with its WT control. Statistical significance of comparisons of these values is given in Table 1.
Dronedarone docking to the hERG channel
Figure 5 shows low energy score configurations for dronedarone (yellow) docked into the open pore of the recently identified, high resolution cryo-EM structure of hERG [27] (Figure 5A) and also to a hERG homology model built on the MthK channel structure (Figure 5B), as this has been found to yield binding configurations that correlate well with experimental data [29, 30]. Dronedarone was able to be docked within the pore of both the MthK model and the hERG open pore cryo-EM structure, providing broadly similar conclusions. In each case low energy configurations placed the protonated aliphatic amino group of dronedarone near the internal binding site for a K+ ion just below the selectivity filter, consistent with competition between drug and K+ ions as observed experimentally. The bulky butyl aliphatic groups on the protonated nitrogen of dronedarone limited the ability of the drug to interact with the side chain hydroxyl groups of S624. In both the cryo-EM structure and homology model, the orientation of the V625 residue suggests that dronedarone would not interact directly with that residue, whilst at least in principle direct interactions with Y652 and F656 could occur.

Figure 5: Low energy score configuration for dronedarone (yellow) docked into the open pore of (A) the high resolution cryoEM structure of hERG and (B) a hERG homology model built on the MthK channel structure. The location of the S5, S6 and pore helices (PH) are indicated on one of the four subunits. The blue star indicates the position of the protonated aliphatic amino group of dronedarone near the binding site for a hydrated K+ ion below the selectivity filter. The purple sphere is a K+ ion in the S3 position of the selectivity filter (a water molecule occupies the S4 position).
Discussion
Relevance of potency of dronedarone inhibition of IhERG
The acute effects of dronedarone on IhERG were first investigated using Xenopus oocyte expression, yielding an IC50 of 9.2 µM at ambient temperature, which is much higher than that subsequently reported from experiments using mammalian cell expression and physiological temperature and the WT IhERG IC50 value in the present study [15, 16, 22]. The disparity between the results from Xenopus and mammalian cell expression in this regard is likely to be due to the presence of vitelline membrane and egg yolk sac in Xenopus oocytes, which can lead to reduced blocking potency when compared to mammalian cells [15, 34]. During clinical administration of 400 mg bid, the maximum plasma values of dronedarone range from 84 to 147 ng/ml (approximately 151 to 264 nM), which exceed the WT IhERG IC50 of 42.6 nM in the present study [35]. Dronedarone has been reported to produce concentration-dependent QT interval prolongation in healthy volunteers and cases of excessive QTc prolongation, ventricular ectopy and Torsades de Pointes have been reported in patients receiving dronedarone [36-38]. In a recent preclinical study of dogs receiving dronedarone, the drug was found to prolong the Tpeak-Tend interval in a dose-related fashion, suggestive of a propensity to prolong transmural dispersion of repolarization and likely to result from IKr inhibition [39]. Thus, the fact that dronedarone is a potent IhERG blocker likely has relevance to both clinical and pathophysiological effects of the drug and it is prudent that the concurrent presence of other risk factors for drug-induced QT interval prolongation should be taken into account when considering administration of this drug [7, 40].
The nature of interactions between dronedarone and the hERG channel
The initial Xenopus oocyte data on IhERG block by dronedarone suggested that dronedarone may interact with closed, open and inactivated hERG channels and revealed some voltage-dependence of inhibition [15]. A subsequent mammalian cell study from our laboratory employed a brief depolarisation protocol to investigate gated versus closed channel block and the results with this were inconsistent with a significant component of closed channel block, instead suggesting rapid development of channel inhibition on channel gating, with no preference for activated over inactivated channels [16]. Similar to the present study, with high [K+]e and inward K+ flux, the extent of dronedarone inhibition of IhERG was reduced [16], consistent with drug binding in or close to the ion conduction pathway and an electrostatic repulsion or “knock off” effect within inward K+ flux [41]. Together with the ability of a profound IhERG blocking concentration (4 µM) of dronedarone to inhibit Y652A and F656A hERG, this led to the possibility that dronedarone might bind relatively high in the channel pore and interact with residues at the base of the pore helices (T623, S624, V625), close to the selectivity filter [16]. Subsequently, we showed that structurally related amiodarone is able to interact with the S624 residue as strongly as with Y652 and more strongly than with F656 [19].
In the present study we tested a dronedarone concentration that was >10-fold the IC50 for outward WT IhERG tail block against pore-helix and S6 mutations. Although statistically significant, the reduction from 97% to 76% inhibition by the Y652A mutation is less extensive than in comparable experiments performed with amiodarone [19], whilst the reduction to 46% for F656A from 75% from its WT control also represents a relatively modest attenuation of block, particularly when considered against other high affinity inhibitors (eg [17, 18, 32, 42]). Unlike for amiodarone [19], however, the S624A mutation produced no significant attenuation of the inhibitory effect of dronedarone, whilst the T623A and V625A mutations produced statistically significant, but relatively modest reductions in block (Table 1) that appear inconsistent with predominant interactions between the drug and pore helical residues near the channel’s selectivity filter. Intriguingly, the mutation with the greatest effect on the IhERG inhibitory effect of dronedarone was V659A (a reduction to 36% inhibition from 82% for its WT control, Figure 4 and Table 1). Indeed, approximation of IC50 values for the different hERG mutations (by inserting into the Hill equation the fractional block values measured at 600 nM and a Hill coefficient of 1.0) suggested that none of the mutations other than V659A would produce a dronedarone IC50 >4.5 fold that for the WT channel, whereas for V659A this would be approximately 8-fold. Interestingly, the L532P mutation in the S4 domain on hERG has been seen to produce an IC50 for dronedarone that is 5.3 fold that for WT IhERG [22]. This residue is too far from the pore cavity to participate directly in binding drugs that interact with canonical binding residues and may exert its effect through an allosteric action in positioning key binding residues [22]. Thus, changes in drug potency of the order of those seen in the present study do not necessarily indicate a direct interaction between the mutated residue and drug binding. However, it is noteworthy that a prior in silico study using a modified KcsA-based hERG channel model found dronedarone to fit well within the channel’s inner cavity with the oxygen atoms of the methanesulphonamide group able to interact with side-chains of T623 and S624 [43]. Our experimental data are compatible with some potential interactions with T623, but not with S624. In that model, dronedarone was able also to interact with Y652 and F656 residues and the authors commented that partial (as opposed to profound) attenuation of block by mutations at these residues [16] might feasibly reflect the ability of one aromatic residue to compensate for the other (i.e. loss of Y652 interactions in the Y652A hERG mutant may be compensated by interactions with F656 and vice versa) [43]. Such compensation has also been suggested for other molecules for which mutation of a single aromatic residue has comparatively little effect on hERG block [21]. Our docking simulations were performed with a MthK based hERG homology model which has been used previously to study hERG binding of a number of drugs and to produce results that accord well with those from experimental data [19-21, 24, 29, 30]. We used this model in addition to the recent hERG structure obtained from cryo-EM [27] because recent data raise the possibility that the hERG structure may have been captured in a conformation that is not optimal for studying high affinity drug binding [30]. In both the MthK model and the hERG open pore cryo-EM structure (Figure 5) low energy configurations placed the protonated aliphatic amino group of dronedarone near the internal binding site for a K+ ion just below the selectivity filter, consistent with competition between drug and K+ ions as observed experimentally. Unlike with amiodarone [19], the bulky butyl aliphatic groups on the protonated nitrogen of dronedarone limit interaction with the side chain hydroxyl groups of S624 and this may explain the absence of effect of S624A on dronedarone block. In neither the MthK model nor the cryoEM structure could dronedarone make direct interactions with the V659 side chain, which is oriented away from the channel pore towards the S5 helix. The moderate (approx. 8-fold) effect of V659A is therefore most likely due to an effect on the configuration of the pore as a result of altered packing at the S5-S6 interface that also results in perturbed channel gating in hERG V659A [44]. The modest effect on channel block in both the Y652A and F656A hERG mutants is more difficult to explain in the context of docking since the drug should come into contact at least with Y652 in the cryo-EM structure and with both Y652 and F656 in the MthK model. As discussed above, one potential explanation for this is reciprocal compensating interactions involving these residues. Accordingly, future work is warranted to determine whether combining mutations at Y652 and F656 is able to eliminate high affinity IhERG block by dronedarone, or whether alternative potential binding residues to those studied here (eg [45]) contribute to the drug’s potent inhibitory action on IhERG.
Conflict of interest
The authors declare that they have no conflicts of interest.
Acknowledgements
The authors thank the British Heart Foundation for financial support (PG/06/42; PG/17/89/33414). JCH also acknowledges receipt of a University of Bristol Research Fellowship.
Article Info
Article Type
Research ArticlePublication history
Received: Thu 26, Jul 2018Accepted: Tue 14, Aug 2018
Published: Fri 31, Aug 2018
Copyright
© 2023 Jules Hancox. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited. Hosting by Science Repository.DOI: 10.31487/j.JICOA.2018.01.002
Author Info
Corresponding Author
Jules HancoxSchool of Physiology, Pharmacology and Neuroscience, Medical Sciences Building, University Walk, University of Bristol, BS8 1TD, United Kingdom
Figures & Tables
References
1. Mitcheson JS, Sanguinetti MC (1999) Biophysical properties and molecular basis of cardiac rapid and slow delayed rectifier K channels. Cell Physiol Biochem 9: 201-216. [Crossref]
2. Sanguinetti MC, Jiang C, Curran ME, Keating MT (1995) A mechanistic link between an inherited and an acquired cardiac arrhythmia: HERG encodes the IKr potassium channel. Cell 81: 299-307. [Crossref]
3. Trudeau MC, Warmke JW, Ganetzky B, Robertson GA (1995) HERG, an inward rectifier in the voltage-gated potassium channel family. Science 269: 92-95. [Crossref]
4. Sanguinetti MC, Tristani-Firouzi M (2006) hERG potassium channels and cardiac arrhythmia. Nature 440: 463-469. [Crossref]
5. Hancox JC, Whittaker DG, Du C, Stuart AG, Zhang H (2018) Emerging therapeutic targets in the short QT syndrome. Expert Opin Ther Targets 22: 439-451. [Crossref]
6. Vandenberg JI, Walker BD, Campbell TJ (2001) HERG K+ channels: friend and foe. Trends Pharmacol Sci 22: 240-246. [Crossref]
7. Hancox JC, McPate MJ, El Harchi A, Zhang YH (2008) The hERG potassium channel and hERG screening for drug-induced torsades de pointes. Pharmacol Ther 119: 118-132. [Crossref]
8. Camm AJ, Kirchhof P, Lip GY, Schotten U, Savelieva I, et al. (2010) Guidelines for the management of atrial fibrillation: The Task Force for the Management of Atrial Fibrillation of the European Society of Cardiology (ESC). Eur Heart J 31: 2369-429. [Crossref]
9. Marinelli A, Capucci A (2012) Amiodarone (Nexterone) injection for the treatment and prophylaxis of frequently recurring ventricular fibrillation. Expert Opin Pharmacother 13: 573-584. [Crossref]
10. Kodama I, Kamiya K, Toyama J (1997) Cellular electropharmacology of amiodarone. Cardiovas Res 35: 13-29. [Crossref]
11. Doggrell SA (2001) Amiodarone - waxed and waned and waxed again. Exp Opin Pharmacother 2: 1-14. [Crossref]
12. Doggrell SA, Hancox JC (2004) Dronedarone: an amiodarone analogue. Exp Opin Invest Drugs 13: 415-426. [Crossref]
13. Varro A, Takacs J, Nemeth M, Hala O, Virag L, Iost N, et al. (2001) Electrophysiological effects of dronedarone (SR 33589), a noniodinated amiodarone derivative in the canine heart: comparison with amiodarone. Br J Pharmacol 133: 625-634. [Crossref]
14. Gautier P, Guillemare E, Marion A, Bertrand JP, Tourneur Y, et al. (2003) Electrophysiological characterization of dronedarone in guinea-pig ventricular cells. J Cardiovas Pharmacol 41: 191-202. [Crossref]
15. Thomas D, Kathofer S, Zhang W, Wu K, Wimmer AB, et al. (2003) Acute effects of dronedarone on both components of the cardiac delayed rectifier K+ current, HERG and KvLQT1/minK potassium channels. Br J Pharmacol 140: 996-1002. [Crossref]
16. Ridley JM, Milnes JT, Witchel HJ, Hancox JC (2004) High affinity HERG K+ channel blockade by the antiarrhythmic agent dronedarone: resistance to mutations of the S6 residues Y652 and F656. Biochem Biophys Res Comm 325: 883-891. [Crossref]
17. Mitcheson JS, Chen J, Lin M, Culberson C, Sanguinetti MC (2000) A structural basis for drug-induced long QT syndrome. Proc Natl Acad Sci U S A 97: 12329-12333.
18. Kamiya K, Niwa R, Mitcheson JS, Sanguinetti MC (2006) Molecular determinants of HERG channel block. Mol Pharmacol 69: 1709-1716. [Crossref]
19. Zhang Y, Colenso CK, El Harchi A, Cheng H, Witchel HJ, et al. (2016) Interactions between amiodarone and the hERG potassium channel pore determined with mutagenesis and in silico docking. Biochem Pharmacol ;113: 24-35. [Crossref]
20. El Harchi A, Zhang YH, Hussein L, Dempsey CE, Hancox JC (2012) Molecular determinants of hERG potassium channel inhibition by disopyramide. J Mol Cell Cardiol 52: 185-195. [Crossref]
21. Du C, Zhang Y, El Harchi A, Dempsey CE, Hancox JC (2014) Ranolazine inhibition of hERG potassium channels: Drug-pore interactions and reduced potency against inactivation mutants. J Mol Cell Cardiol 74: 220-230. [Crossref]
22. Zhang YH, Colenso CK, Sessions RB, Dempsey CE, Hancox JC (2011) The hERG K+ channel S4 domain L532P mutation: characterization at 37 degrees C. Biochim Biophys Acta 1808: 2477-2487. [Crossref]
23. Milnes JT, Crociani O, Arcangeli A, Hancox JC, Witchel HJ (2003) Blockade of HERG potassium currents by fluvoxamine: incomplete attenuation by S6 mutations at F656 or Y652. Br J Pharmacol 139: 887-898. [Crossref]
24. Melgari D, Brack KE, Zhang C, Zhang Y, El Harchi A, et al. (2015) hERG potassium channel blockade by the HCN channel inhibitor bradycardic agent ivabradine. J Am Heart Assoc pii e001813. [Crossref]
25. Melgari D, Zhang Y, El Harchi A, Dempsey CE, Hancox JC (2015) Molecular basis of hERG potassium channel blockade by the class Ic antiarrhythmic flecainide. J Mol Cell Cardiol 86: 42-53. [Crossref]
26. Zhang YH, Cheng H, Alexeenko VA, Dempsey CE, Hancox JC (2010) Characterization of recombinant hERG K+ channel inhibition by the active metabolite of amiodarone desethyl-amiodarone. J Electrocardiol 43: 440-448. [Crossref]
27. Wang W, MacKinnon R (2017) Cryo-EM Structure of the Open Human Ether-a-go-go-Related K+ Channel hERG. Cell 169: 422-430. [Crossref]
28. Jiang Y, Lee A, Chen J, Cadene M, Chait BT, et al. (2002) Crystal structure and mechanism of a calcium-gated potassium channel. Nature 417: 515-522. [Crossref]
29. Dempsey CE, Wright D, Colenso CK, Sessions RB, Hancox JC (2014) Assessing HERG pore models as templates for drug docking using published experimental constraints: the inactivated state in the context of drug block. J Chem Inf Model 54: 601-612. [Crossref]
30. Helliwell MV, Zhang Y, El Harchi A, Du C, Hancox JC, et al. (2018) Structural implications of hERG K+ channel block by a high affinity minimally-structured blocker. J Biol Chem 293:7040-7057. [Crossref]
31. Mitcheson JS, Chen J, Sanguinetti MC (2000) Trapping of a methanesufonanlide by closure of the HERG potassium channel activation gate. J Gen Physiol 115: 229-240. [Crossref]
32. Kamiya K, Niwa R, Morishima M, Honjo H, Sanguinetti MC (2008) Molecular determinants of HERG channel block by terfenadine and cisapride. J Pharmacol Sci 108: 301-307. [Crossref]
33. Perry M, Stansfeld PJ, Leaney J, Wood C, de Groot MJ, et al. (2006) Drug binding interactions in the inner cavity of HERG channels: molecular insights from structure-activity relationships of clofilium and ibutilide analogs. Mol Pharmacol 69: 509-519. [Crossref]
34. Witchel HJ, Milnes JT, Mitcheson JS, Hancox JC (2002) Troubleshooting problems with in vitro screening of drugs for QT interval prolongation using K+ channels expressed in mammalian cell lines and Xenopus oocytes. J Pharmacol Toxicol Methods 48: 65-80. [Crossref]
35. Dorian P (2010) Clinical pharmacology of dronedarone: implications for the therapy of atrial fibrillation. J Cardiovasc Pharmacol Ther 15: 15S-8S. [Crossref]
36. Wadhani N, Sarma JS, Singh BN, Radzik D, Gaud C (2006) Dose-dependent effects of oral dronedarone on the circadian variation of RR and QT intervals in healthy subjects: implications for antiarrhythmic actions. J Cardiovasc Pharmacol Ther 11: 184-190. [Crossref]
37. Gonzalez JE, Sauer WH, Krantz MJ (2013) Ventricular ectopy and QTc-interval prolongation associated with dronedarone therapy. Pharmacotherapy 33: e179-e181. [Crossref]
38. Huemer M, Sarganas G, Bronder E, Klimpel A, Garbe E, et al. (2015) Torsade de pointes tachycardia in a patient on dronedarone therapy. Pharmacotherapy 35: e61-e65. [Crossref]
39. Motokawa Y, Nakamura Y, Hagiwara-Nagasawa M, Goto A, Chiba K, et al. (2018) In vivo Analysis of the Anti-atrial Fibrillatory, Proarrhythmic and Cardiodepressive Profiles of Dronedarone as a Guide for Safety Pharmacological Evaluation of Antiarrhythmic Drugs. Cardiovasc Toxicol 18: 242-251. [Crossref]
40. Yap YG, Camm AJ (2003) Drug induced QT prolongation and torsades de pointes. HEART (89):1363-1372.
41. Wang S, Morales MJ, Liu S, Strauss HC, Rasmusson RL (1997) Modulation of HERG affinity for E-4031 by [K+]o and C-type inactivation. FEBS Lett 417: 43-47. [Crossref]
42. Ridley JM, Witchel HJ, Hancox JC (2006) Clemastine, a conventional antihistamine, is a high potency inhibitor of the HERG K+ channel. J Mol Cell Cardiol 40: 107-118. [Crossref]
43. Stansfeld PJ, Gedeck P, Gosling M, Cox B, Mitcheson JS (2007) Drug block of the hERG potassium channel: insight from modeling. Proteins 68: 568-580. [Crossref]
44. Mitcheson JS (2008) hERG potassium channels and the structural basis of drug-induced arrhythmias. Chem Res Toxicol 21: 1005-1010. [Crossref]
45. Saxena P, Zangerl-Plessl EM, Linder T, Windisch A, Hohaus A, et al. (2016) New potential binding determinant for hERG channel inhibitors. Sci Rep 6: 24182.