Understanding Checkpoint Inhibitors in Cancer Therapy, Mechanisms of Action, Resistance and Future Challenges
A B S T R A C T
The immune system is the human body’s natural defence against mutated cells produced as the result of DNA replicative error or by the effect of carcinogens, a process rereferred to as immune surveillance. ‘Escaping’ of cancer cells from immune surveillance leads to tumor development, metastasis and progression. Avoiding detection and destruction by the immune system are the result of cancer cells evolution, caused primarily by cancer cells’ genomic instability. On the other hand, scientists attempted for decades to exploit the anticancer effect of the immune system with limited success. However, better understanding of the mechanisms behind the cancer cells’ ability to avoid detection and suppression by the immune system resulted in the development of immune checkpoint inhibitors, a form of immunotherapy, first approved by the Food and Drug Administration (FDA) in 2011. This article reviews the pathways involved in anticancer immune response, evading and supressing of the immune system by cancer cells mechanisms of action and successes of immune checkpoint inhibitors (ICI), particularly programmed death-1 (PD-1) and programmed death-ligand (PD-L1) inhibitors as well as mechanisms that result in resistance of cancer cells to ICI.
Keywords
Immune check point inhibitors, resistance to immune check point, inhibitors, mechanism of action of immune check point inhibitors, tumor microenvironment, cancer immunotherapy, cancer immunology
Introduction
Paul Ehrlich theorised that immune system has a crucial role in detecting and eliminating cancer cells more than 100 years ago [1]. “Non targeted forms of immunotherapy” such as interferon-alpha (IFN-a) and interleukin-2 (IL2) have been in clinical use for years, with variable level of success, in metastatic renal cells carcinoma and melanoma [2].The non-specific overstimulation of the immune system by IFN-a and IL2, has the potential to cause serious and sometimes life threatening (such as cytokine storm) complications, as the result, the use of these therapies has been limited to highly selective and otherwise fit patients [3]. Better understanding of the complex relationship between cancer cells and their ecosystem, otherwise known as tumor microenvironment (TME), provided insight into the mechanisms involved in cancer cells’ ability of evasion of detection and elimination by immune system [4, 5].
The emergence of ICIs to unleash the cytotoxic effect of the immune system has been the product of a greater insight into the intricate interaction between cancer cells, TME and the immune system. As ICIs exert their effect through in a more specific mechanisms compared to older versions of immunotherapy, they possess better safety, side effect profile and better tolerance by patients [6]. An important challenge in clinical practice, is the tolerance that cancer cells develop during the course of the disease to chemotherapy through a number of resistance mechanisms. As monoclonal antibodies, ICIs, are not affected by these mechanisms [7]. Therefore, providing a crucial therapeutic line against cancer.
Immune Surveillance and Cancer Cells Destruction
Both innate and adaptive components of the immune system play crucial roles in cancer cell detection and destruction. Cytotoxic lymphocytes (CL), cytotoxic T cells (CD4 +ve CD8 +ve T-cells), and natural killer (NK) cells, are key players. Dendritic cells (DC) capture antigens (Ag) on the cell surface of cancer cells. Activation of DC is required to adequately trigger cytotoxic T-cells activation and maturation [5, 6]. Danger signals known damage associated molecular patterns (DAMPS) are activators of DC. DAMPS are produced by cells in distress such as cancer cells or viral infected cells. Of note, not all cancer cells release DAMPS since it is a mechanism deployed by cancer cells to avoid immune detection and destruction [8]. DCs then present these Ag in the context of major histocompatibility complex (MHC) molecules to T lymphocytes within lymph nodes (Figure 1) [8].
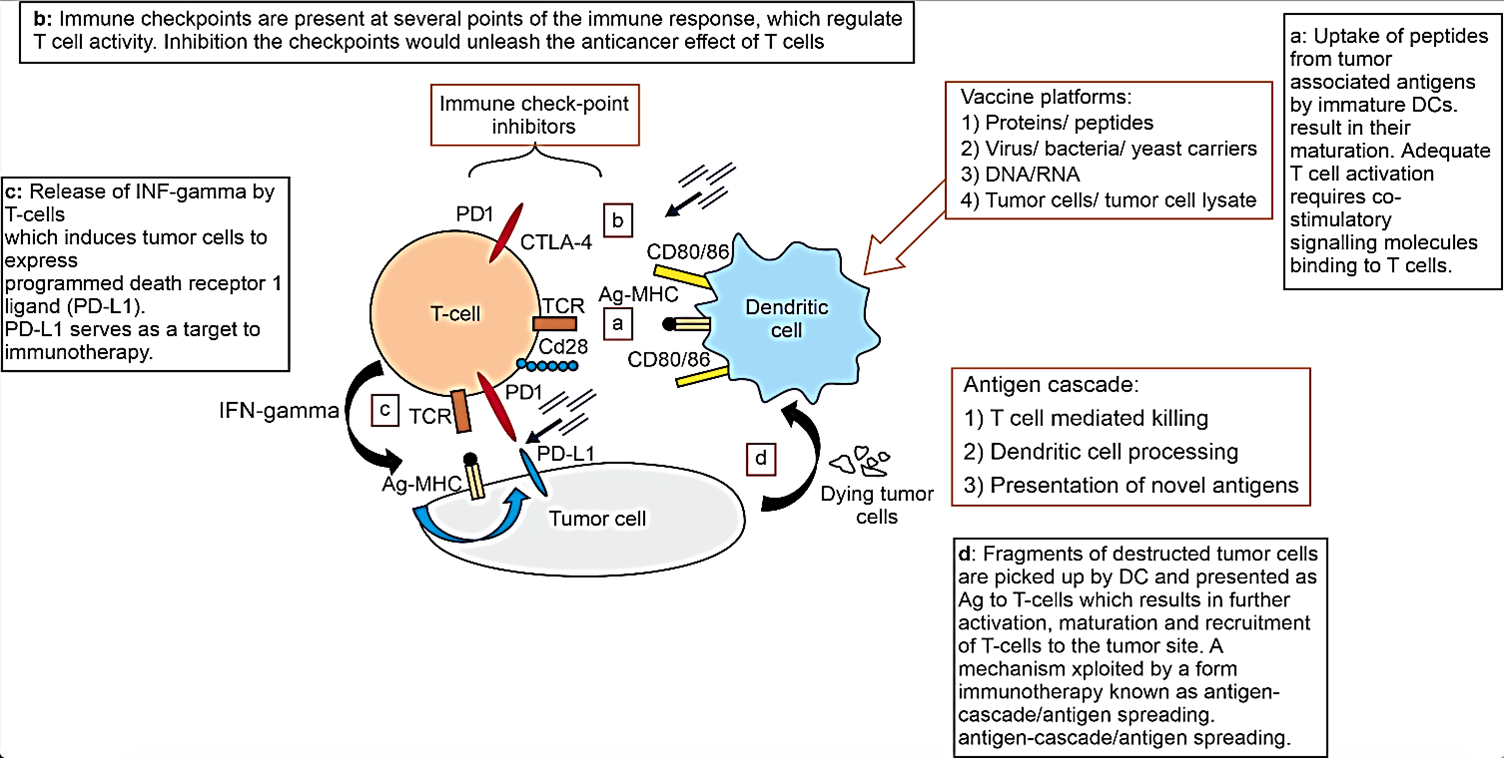
Figure 1: An illustration of the complex interaction between DC, T-cells and tumor cells. Also showing potential targets for immunotherapy.
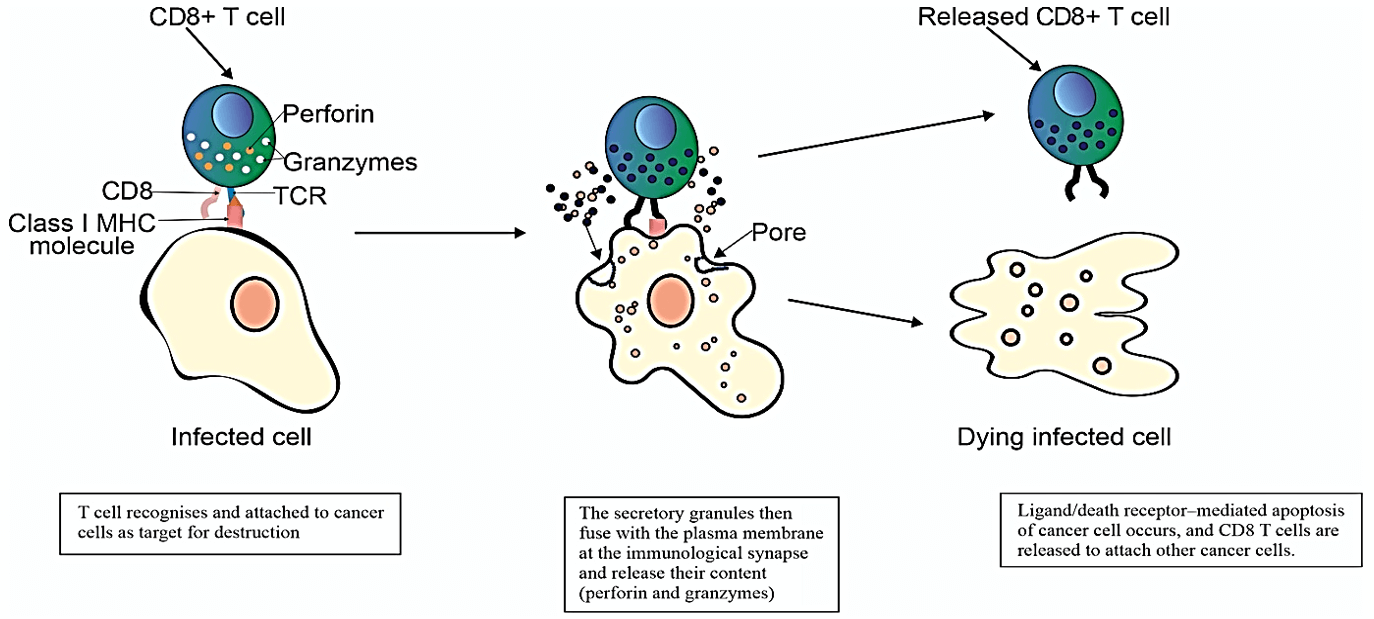
Figure 2: Illustration of anticancer effect of cytotoxic T-cells upon exposure to tumor specific antigen presented by DC.
This occurs by translocating MHC from cytoplasm to the cells surface. Upon activation of cytotoxic T-cells on exposure to Ag presented by DC, T-cells undergo proliferation, maturation and arrive at tumor site to exert their cytotoxic effect on the cancer cells (Figure 2). Activation of CTL requires presenting of neoantigen (MHC) by the antigen presenting cells (APC) via MHC to T cell receptor (TCR) on the surface of CTL. This activation is only adequate if positive co-stimulatory signalling occurs via secondary co-stimulatory receptors and ligands, the best characterised are B7 on the surface of APC and CD28 on the surface CD4 and CD8 CTL. Lack of secondary co-stimulation would lead to CTL anergy and tolerance [7].
A third crucial element of adequate activation of CTL by DC is the release of stimulatory cytokines IL-12, IL-2 and INF gamma. Failure of the release of these cytokines will also leads to CTL anergy and tolerance [9]. In addition, there are a number of co-inhibitory receptors and ligands which are responsible of truncating the immune system to avoid autoimmunity. Co-stimulatory and co-inhibitory ligands and their receptors on the surfaces of antigen presenting cells (APC) and T cells respectively, work in synchrony to regulate the cytotoxic effect of the T cells. The temporal relation between the activatory and inhibitory pathways determines the function of the T-cells according to the physiological need. Potentiation and inhibition of both co-stimulatory and co-inhibitory ligands respectively, have been explored to exert anticancer therapeutic effect with mixed outcomes. For example, the experiment on six healthy human volunteers of a CD28 superagonist antibody named TGN1412 resulted in disastrous complications, as all the volunteers suffered multiorgan failure caused by cytokine storm and ended up in intensive care [10].
Cancer immunoediting, a term describes attenuating anti-tumor immune responses, refers to the process of progressing from immune elimination of cancer, to a state of equilibrium (typically temporary state) to a phase of cancer escaping the immune system where it becomes clinically detectable. In the elimination phase the immune system recognise the nascent cells and destroy them before they develop to malignancy. During the equilibrium phase the cancer cells are dormant and controlled by the immune system in a state of equilibrium, often temporarily, but not fully eliminated, albeit some cancer cells acquire the capability of evading the immune system. In the escape phase, the immune system is overrun by the cancer cells and progress to clinically detectable tumors.
The release of immunosuppressive factors, harnessing an immunosuppressing tumor microenvironment and the formation of physical barriers (tumor stroma) which prevents the infiltration of tumor bedding by cytotoxic T cells, represent a key event during the escape phase [11].
CTL deploy two main mechanisms in killing cancer cells; one is evoked by granule exocytosis i.e. perforin (PRF1) and granule-associated enzymes (granzymes; GZM)] [12]. Serine proteases are the major constituents of the cytotoxic granules and are very toxic to cancer cells [12]. Once GZM released into the cancer cells, they cleave substrates crucial for cancer cell survival. The other mechanism is through the death ligand/death receptor system. Upon induction of T-cell receptor (TCR) or through killer activating receptors (KAR), effector cells release death ligands like FasL (Fas ligand) and TRAIL (TNF-related apoptosis inducing ligand) [10]. Both of these mechanisms lead to induction of intracellular apoptotic pathway in the cancer cells [8]. However, there is evidence that cancer death might occur even in the presence of antiapoptotic factors i.e. inhibitors of apoptosis (IAP) or Bcl-2 (B-cell lymphoma) family members; which means CL-mediated killing might involve non-apoptotic pathways too [13, 14]. Furthermore, studies from xenograft models demonstrated a central of PRF1-mediated cell death. PRF1-knocked out mice had impaired cell death, develop spontaneous B lymphoma and accelerates the onset of HER2/neu-driven breast carcinomas [15, 16].
Immune Escape Mechanisms by Cancer Cells
Evading immune detection and elimination by cancer cells involves complex mechanisms (Figure 3). Cancer cells maintaining tumor specific Ag (TSAg) represent a key step in the immune system’s ability to discern cancer cells from normal cells, this is referred to as cancer antigenicity [17]. Viral derived proteins, proteins encoded by cancer-germline genes, differentiation antigens, and proteins arising from somatic mutations or gene rearrangements are examples of well-studied TSAg [17].
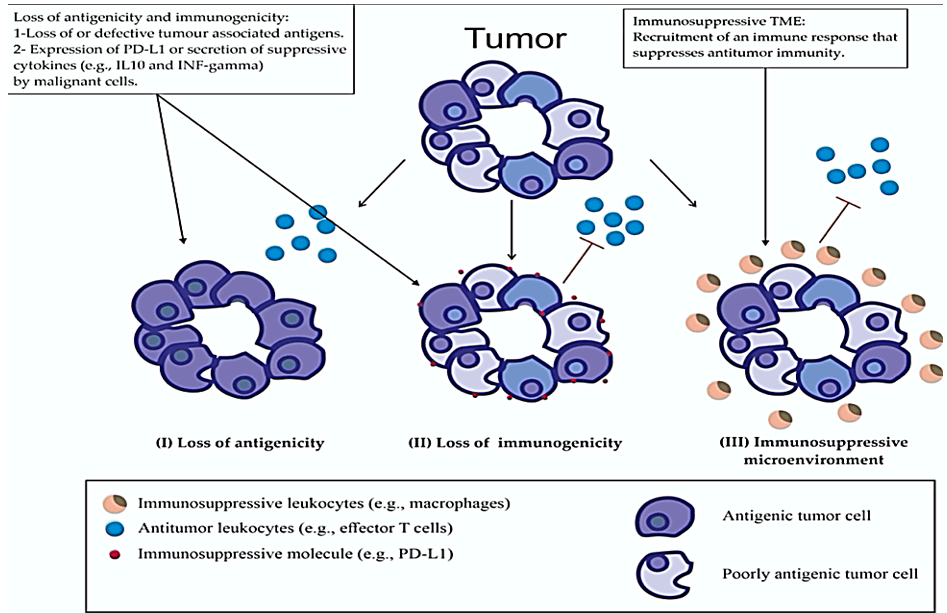
Figure 3: Mechanisms involved in cancer cells evading immune detection and elimination.
Due to the intrinsic genomic instability, cancer cells can loss their antigenicity, due to the loss of or acquisition of immature, defective or mutated TSAg. Loss of MHC expression or defective Ag processing machinery are examples of loss of antigenicity [17]. Although, inter and intra tumor heterogeneity result in a diverse range of TSAg, which at least in theory, expands the opportunity to therapeutically exploit the immune system [18]. Immunogenicity of cancer cells might be predicted by rate of mutation, for example melanoma is known to possess high mutation rate and response to immunotherapy. However, other cancers such as renal cell and bladder cancer respond well to immunotherapy despite their low mutation rate [19]. Loss of adequate MHC class 1 expression or defective Ag presentation by malignant cells to cytotoxic T cell via DC would impair detection and subsequent destruction by immune system. MHC class 1 molecules under-expression has been detected in about 20-60% of melanoma, lung, breast, renal, prostate, and bladder cancers [20]. Another immunosuppressive mechanism is the release of immunoinhibitory cytokines such as IFN-gamma by tumor-infiltrating lymphocytes. IFN-gamma upregulates PD-L1 on the malignant cell surface [20, 21]. An immunosuppressive TME allows cancer cells to avoid immune destruction. Such TME is often seen to be heavily infiltrated with immune inhibitory inflammatory cells, such as macrophages, that oppose the cytotoxic effect of T-cells [22].
The Role of the Immunosuppressive Tumor Microenvironment (TME)
Despite the growing understanding of immunosuppressive TME in the recent years, more research is required to untangle the intricate relationship between cancer cell and its ecosystem. Cancer cells are able through secreting specific cytokines and chemokines to recruit a range of immunosuppressant cells to its microenvironment (Figure 4).
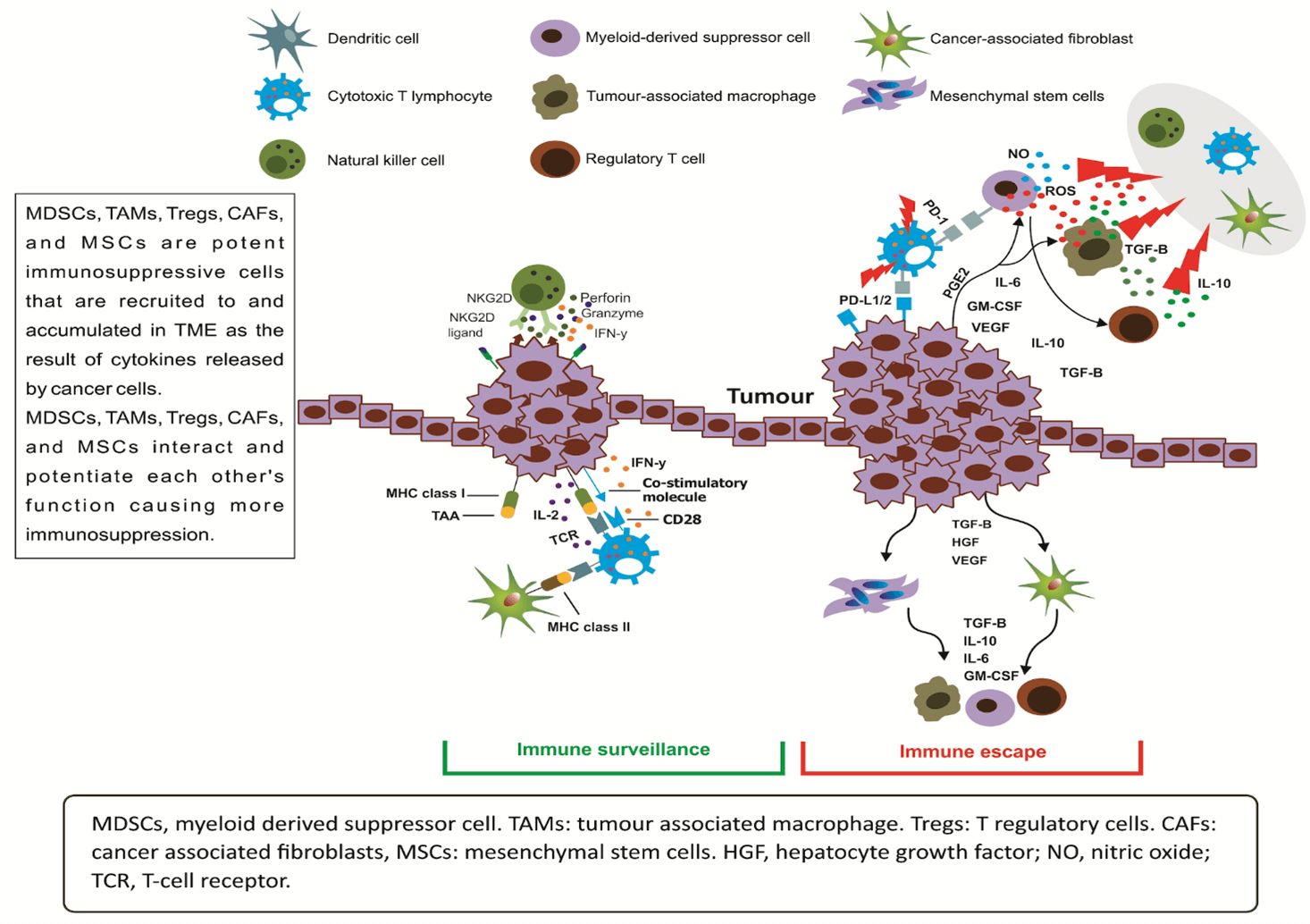
Figure 4: The immunosuppressive tumor microenvironment (TME) support the growth of tumor through exerting its anti-immune effect via recruiting immunosuppressing cells and secreting immune inhibitory cytokines.
Suppressor T and tumor cells secrete a complex network of protumor cytokines that interact with immune cells to promote tumor growth and progression. Important immune supressing factors secreted by tumor cells and their functions include: IL-10 which inhibits the function of APC, IL-12 inhibits B7 expression, transforming growth factor beta (TGF-beta) inhibits T cells proliferation, IL-4 inhibits the action of interferon gamma, IL-10 and TGF-beta both inhibit macrophage activation [23]. Myeloid‐derived suppressor cells (MDSCs), contribute to immunosuppressive TME and represent a heterogeneous population of immature myeloid cells [21]. The generation of MDSCs in and departure from the bone marrow and deposition at TME are driven by the release by cancer cells of proinflammatory cytokines, growth and chemotactic factors. Studies of cancer xenograft models and cancer patients showed MDSCs exert their inhibitory effect on cytotoxic T cells through the release of arginase 1 (ARG1), indoleamine‐2, 3‐dioxygenase (IDO), inducible nitric oxide synthase (iNOS) and reactive oxygen species (ROS) [24].
MDSCs’ further contribution to the immunosuppressive TME is by potentiating the anti-immune effect of tumor associated macrophages (TAMs) and T regulatory cells (Tregs) [25, 26]. The exact demarcation between MDSCs and TAM is not very clear [27]. They carry immunosuppressive genes (ARG1 and iNOS) and possess potent T‐cell‐suppressive capacity [25]. Another link between MDSCs and TAMs is through sharing same myeloid lineage as monocyte‐derived MDSCs (mo‐MDSCs) [28]. A separating feature between MDSCs and TAMs is that TAMs show low‐to‐intermediate expression of Ly6C and upregulated F4/80 and interferon regulatory factor 8 [29]. Normally Tregs are responsible for regulating the cytotoxic effects of CD4CD8 CTL. Indeed, inactivation of Tregs is thought be involved in many autoimmune conditions. In tumors, TGF‐β and IL‐10 cause activation of Tregs and their infiltration the tumor stroma. This in turn leads to suppression of (and therefore immunosuppression) CDs and other APCs and CD4CD8 CTL [30].
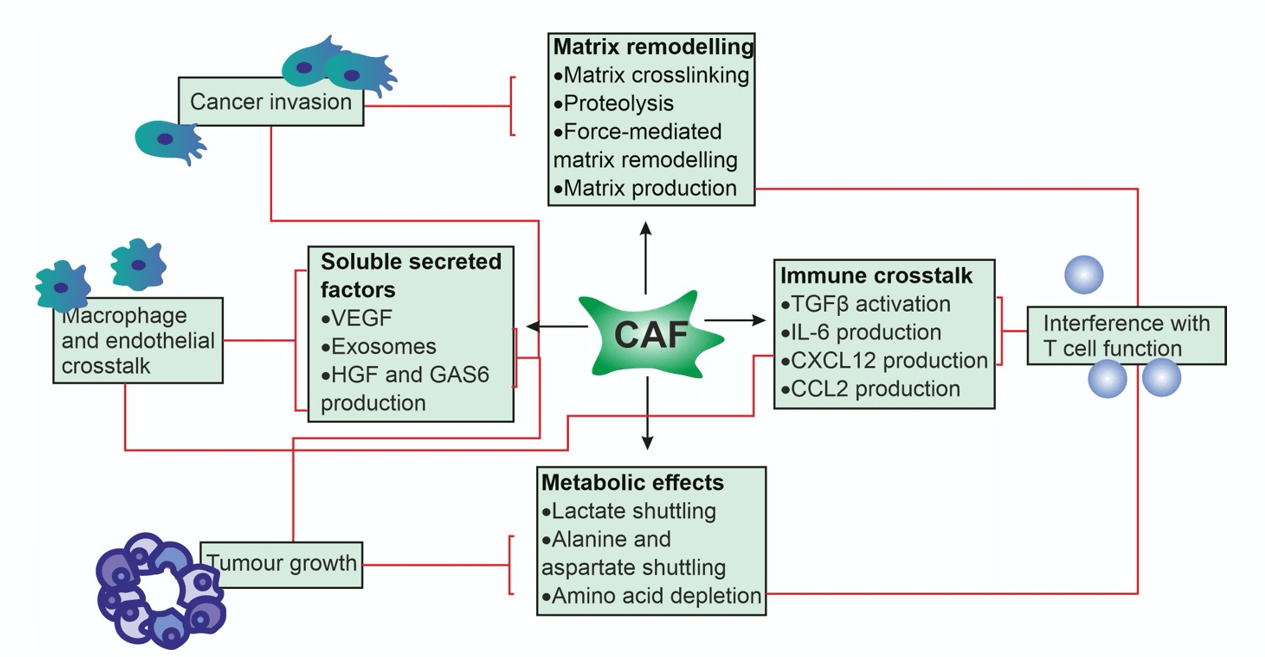
Figure 5: The effect of CAF on the TME is either direct via production of growth factors and cytokines and mitochondrial fuel (e.g. lactate, fatty acids) for cancer cell metabolism or indirect via promoting angiogenesis through secretion of pro-angiogenic factors (e.g. FGF2, VEGF, SDF-1) and remodeling of TME to a more protumor state.
An important component of TME is Cancer-associated fibroblasts (CAFs), which have a complicated range of effects on TME (Figure 5) [28]. CAFs usually, but not always, express α-smooth muscle actin (αSMA), Fibroblast Activation Protein (FAP), and Platelet Derived Growth Factor Receptor β (PDGFRβ). Compared to normal fibroblasts CAFs, are phenotypically different which are characterised by high motility and metabolic activity and high proliferative state [31]. Moreover, CAF promote tumor invasion through direct degradation and remodelling of extra-cellular matrix [31]. The immunosuppressing effects of CAF are the result of interference with the function of CTL through activation of TGF-beta by production of CCL2, CC-chemokine ligand 2; CXCL12, CXC-chemokine ligand 12 by CAF [31, 32].
Dysregulation of DC is believed to be another contributor to an anti-immune TME. One postulated mechanism is the inhibition of the maturation of antigen presentation function of DCs by aberrant expression of miRNAs such as miR‐22, miR‐146a and miR‐146b which leads to failure of activation of CTL [33]. Another mechanism is preventing maturation (hence defective function) of DCs by TGF‐β through induction of global H3K4me3 and H3K27me3. TGF‐β is known to be abundant in most immunosuppressive TMEs [34]. Transgenic mice models with knock of genes that lead to defective T‐cell antigen sensitivity, showed the importance of epigenetic changes that can adversely affect adequate functionating of CD4CD8 CTL [35].
Immune Checkpoint Inhibitors (ICI)
I Proof of Efficacy and Safety
This class of immunotherapy proven to be effective in the treatment of important metastatic malignancies such as non-small cell lung cancer, melanoma renal cell carcinoma, bladder cancer, MSI-high colorectal carcinoma, Merkel cell carcinoma, head and neck squamous cell carcinoma, and Hodgkin lymphoma [36]. In one study, 20% of melanoma patients (a highly mutated and immunogenic malignancy) treated with Ipilimumab (anti-CTLA-4) achieved durable long-term (5 to 10 years) response [37]. Treatment of melanoma with Pembrolizumab (anti-PD-1) resulted in 70-80% initial and 33% response at 3 years [38]. Treating metastatic melanoma with anti-CTLA-4 and anti-PD-1 combination showed even greater response rate (RR 58%), albeit 50% of patients experienced significant adverse events, including grade 4 pneumotits and colitis [39]. Moreover, one metanalysis concluded that compared to younger adults PD-1 (Nivolumab and Pembrolizumab) and PD-L1 (Atezolizumab) inhibitors had comparable efficacy in adults with metastatic cancer in people older than 65 years of age [36]. Furthermore, two large meta-analysis investing the impact of age on the efficacy of ICIs used for treatment of multiple metastatic cancer types showed no difference between young and older patients (cut off = 65 years) [40, 41]. In addition, a systematic review of 23 randomized controlled trials involving 9322 men (67.9%) and 4399 women (32.1%) with advanced cancer receiving ICIs showed no statistically significant impact of patients’ gender on the efficacy of ICIs measuring overall survival as the primary outcome [42].
In general, concerns regarding side effects caused by autoimmune response caused by the use of ICIs are usually offset by their significant benefits. Albeit, important side effects such as severe colitis, pneumonitis or myocarditis can be considered as life threatening outcomes which warrant permanent discontinuation of ICIs. Due to their pharmacodynamic and pharmacokinetic, unlike chemotherapy, ICIs cause delayed and prolonged immune-related adverse events (irAES) [43]. The core mechanism of irAES of ICIs is the ability of this class of drugs to unleash cellular immunity and disruption to immunologic self-tolerance [44]. ICIs also stimulate B cells to produce autoantibodies which contribute to the development of irAES [45].
Inhibitors of ctla-4 tend to cause more irAES in comparison to PD-1 and PD-L1 inhibitors. This is because ctla-4 inhibitors cause higher generalised and non-specific activation to naïve and memory T cells within lymph nodes [46]. However, PD-1 and PD-L1 inhibitors exert the effects on T-cells within end tissues such as the lung, gastrointestinal, thyroid, skin or the liver [46]. This difference in the irAES between CTLA-4 and PD-1 blockade are consistent with data from murine deficient models CTLA-4 and PD-1 [45]. Deficiency of CTLA-4 in mince is rapidly lethal and precipitate with early onset of progressive lymphoproliferative disorders with infiltration of multiple organs by polyclonal T cells, whereas PD-1 knocked out mice had near normal life span develop a more insidious and slowly progressive autoimmune disorders such as rheumatoid arthritis and dilated cardiomyopathy [47-49].
Not surprisingly, it has been noted that frequency of irAES correlate with the anti-tumor efficacy of ICIs. A metanalysis of 30 studies involving 4971 patients demonstrated a correlation between benefit in overall survival (OS), progression free survival (PFS) and irAES. Low-grade irAES and irAES affecting skin and endocrine organs are particularly correlated with more driven benefit from ICIs [50]. While patients with pre-existing autoimmune diseases are at a higher risk of developing irAES and flare up of their autoimmune conditions, irAES are mostly manageable without the need for permanent discontinuation of ICIs [51].
II Mechanism of Action of ICIs and Biomarkers
Although our knowledge of ICIs continues to grow, the exact mechanisms of action of ICIs are not fully understood. This is partly because of having limited immune competent pre-clinical models showing cancer response to ICIs and partly because of an incomplete insight to other factors (clinical, molecular, and immunologic) that predict response to ICI [52]. Compared to CTLA-4, PD-1/PD-L1 blockade appears to be safer due to less frequent off target action and more effective, as PD-1 is more expressed than CTLA-4. Another characteristic of PD-1 is that chronic antigen exposure (e.g. chronic viral infection) causes persistently high levels of PD-1 which in turn result in T-cell exhaustion [53, 54]. PD-1 blockade also increases the lytic activity of NK cells [38, 54]. As PD-1 expressed in tumor-infiltrating lymphocytes (TILs) from many different tumor types, their blockade enhances the anti-tambour activity within TME. PD-1/PD-L1 blockade leads to reactivation and clonal-proliferation of primed CTL (CTL priming occurs through presenting tumor associated antigens by APCs to CD4CD8 CTL) within the TME [40].
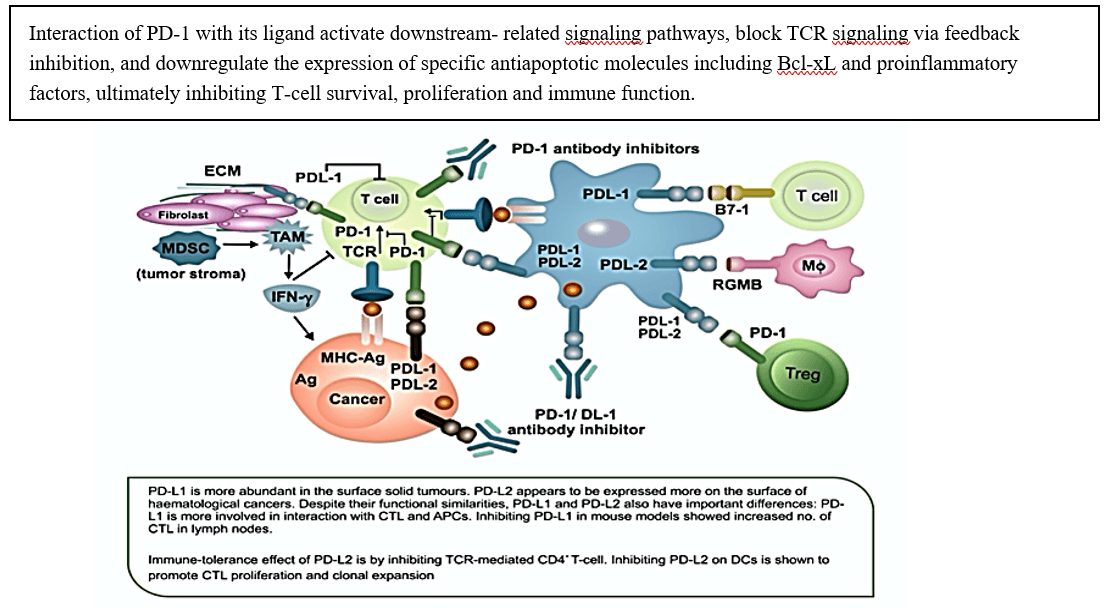
Figure 6: Depiction of the anti-immune effect of the interaction between PD-1 and its ligands. and immune destruction of tumor by ICI is through preventing the binding of PD-1 to its ligands.
A durable response to ICIs requires differentiation of primed CTL into effector memory T cells (TEM) [55]. In normal cells PD-1 (present on CTL, NK-TC, DC and Tregs) responsible to “put a break” to attacking of normal cells by the immune system. Immune cells recognise and stop attaching ‘normal’ cells via the interaction between PD-1 and PD-L1. PD-L1 expressed on epithelial, endothelial, hematopoietic and tumor cells. PD-L2, present on TAM, activated DCs and MDSCs, and has negative effect on CTL, another element of immune tolerance to tumor (Figure 6) [56]. The interaction of PD-1 with its ligands induce tumor-specific T-cell apoptosis and promote the differentiation of cells into Tregs by stopping T cells from entering the G1 phase by upregulation of p15 and reducing the transcription level of SKP2 [57, 58]. Figure 7 shows the effect of cancer cells on the adaptive and innate immune system to avoid immune destruction.
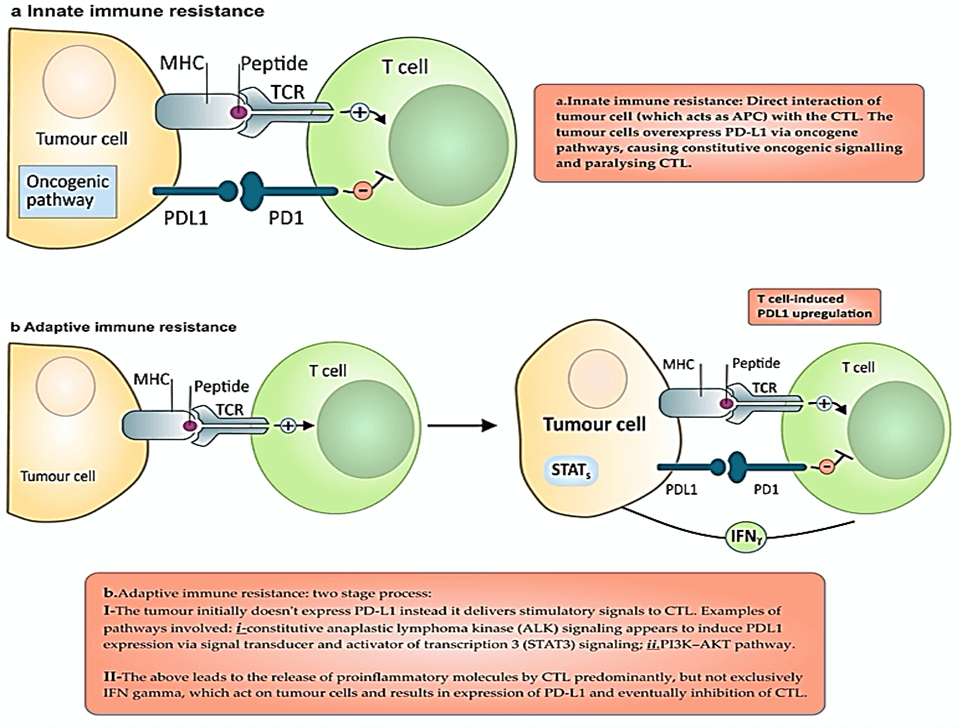
Figure 7: Depiction of two PD-1 and PD-L1 (innate immune resistance vs. adaptive immune resistance) based mechanisms exploited by tumor cells to exert immune tolerance.
Apart from anti PD-1, which is IgG4, almost all the ICIs are either humanized or human immunoglobulin (Ig) G1 antibodies, as such they have similar PD to other monoclonal antibodies with no significant effect on renal and liver function impairment [59]. A steady plasma concentration of ICIs is possible due to their limited diffusion to extravascular space, receptor mediated excretion and prolong half-life [60].
Numerous studies have been conducted to produce sensitive, specific and reliable biomarker that can predict efficacy of ICIs with unwanted side effects and complications. More studies are required to identify precise, none invasive, biological markers that predict response of specific tumor primaries to PD-1/PD-L1 blockade and to design effective and safe therapies to combat immunosuppressive TME, as a hostile cancer ecosystem, which represents one of the main hindrances to response to ICI and predict worse outcome. Having biomarkers that can accurately predict efficacy and irAES would help designing a more rational use of ICIs in appropriate cases with less risk of developing irAES. There are several studies investigating predictive biomarkers of a range of biological characteristics of cancer cells and the TME [61]. Such include the types and number of immune cell infiltrating TME, PD-L1 overexpression, neoantigen clonality, mutational burden, epigenetic factors, mismatch repair and transcription factors [61].
Both immunohistochemistry (IHC) and flow cytometry are the most commonly used techniques to identify biomarkers. Thus far measuring the expression of PD-L1 in tumor cells using IHC is best known biomarker [62]. For example, there is a direct correlation between PD-L1 positivity in melanoma and overall response rate [63]. However, such correlation has not been established with ipilimumab (anti-CTLA antibody) and nivolumab (anti-PD-1 antibody) [64]. The same correlation, on the hand, has been found in other common tumors such as non-small cell lung cancer (NSCLC), colon, rectal and prostate cancer [19]. High mutation burden, measured by DNA-based markers, also correlates well with response to ICIs. Rizvi et al. showed that a higher nonsynonymous mutational burden of NSCLCs predicted improved PFS in response to treatment with anti-PD-1 therapy [65]. In addition, Lipson et al. found hotspot mutations in BRAF, cKIT, NRAS, and TERT identified in circulating tumor cells DNA in patients with malignant melanoma treated with ICIs associated with clinical and radiological tumor progression [66]. Moreover, studies showed increased T cell receptor repertoires predicted an increased immune response in patients receiving radiation, anti-PD-L1 andanti-CTLA-4 therapy [67].
III Isotype of Monoclonal Antibodies and Binding Sites
Antibodies (Abs.) recognise and bind with specific antigens (Ag). This specific relationship allowed treatment with Abs. efficacious with few side effects. Benefiting from this property of Abs., scientists chose IgG4 because of its unique properties, especially in relation to lack of antibody-dependent cytotoxicity (ADCC) and complement-dependent cytotoxicity (CDC), hence not depleting CTL and manufacturing considerations, to make Nivolumab and Pembrolizumab [68]. For PD-L1 inhibitors, IgG1 isotype, which deplete the number of the target (cancer) cell via ADCC, is used [69, 70]. However, Atezolizumab and Durvalumab are engineered to eliminate ADCC to preserve PD-1 and PD-L1 interactions [44]. Avelumab, on the other hand, engineered to utilise immune checkpoint inhibition and ADCC-mediated cytotoxicity of cancer cells synergistically [46]. On binding the antibody to its corresponding ligand/receptor, a stable conformation is formed. This inhibits of binding of PD-1 to its ligands and the therefore the inhibitory signalling and downstream signalling to CTL. The fully human or humanized monoclonal antibodies PD-1 inhibitors (Nivolumab, Pembrolizumab) and PD-L1 inhibitors (Atezolizumab, Durvalumab, Avelumab) are structurally different at antigen epitopes and conformational changes between each inhibitor/molecule and acting domains [49, 71].
IV Resistance to PD-1 and PD-L1 Blockade
Primary resistance describes tumors that have not shown clinical response or stabilized with PD-1/PD-L1 inhibition [52]. Acquired resistance, on the other hand, develops whilst the patient receives or on resumption of PD-1/PD-L1 blockade [50]. To improve therapeutic efficacy, detailed understanding of these complex and often overlapping mechanisms is required. Mechanisms of resistance in each patient might be unique to the patient and likely has been shaped by genetics, patients’ individual tumor characteristics and previous treatment. For instance, history of treatment with chemotherapy might have deleterious effect on patients’ immunity which might in turn contribute to a state of cancer cell tolerance to ICIs [72]. Absence of infiltrating CTL within the tumor, due to inadequate tumor antigenicity, is one of other proposed mechanisms of resistance [73-75]. Pancreatic and prostate cancers are shown to be inherently resistant to PD-1/PD-L1 blockade, because of low rate of somatic mutation which render them less recognisable by the immune system. To tackle inherited tumor deficient antigenicity, infusion of large quantities of CD8+ T cells directed at specific tumor antigens, such as MART-1 in melanoma and NY-ESO-1 in sarcoma, into patients is used; this approach is called adoptive cell therapy (ACT) [76]. Another approach is the expansion and transfusion of peripheral mononuclear blood cells modified via transduction with a TCR directed against a given tumor antigen. Inducing dendritic cells to stimulate tumor antigen specific T-cell activity, via using tumor vaccine, has been shown to be effective [77].
A study by Ribas et al. of melanoma in a phase 1b clinical trial demonstrated that intratumorally injected modified human herpes simplex virus with systemic anti-PD-1 therapy resulted in intensified T cell infiltration in virus-injected lesions that lead to 62% objective response rate, as well as 33% complete response rate [37]. Studies of MHC dysfunctional models of lymphoma and melanoma used of agents that boost MHC expression by epigenetic modification through demethylating and histone deacetylation showed better tumor infiltration by CD8+ T cells and therefore tumor shrinkage [78]. This study confirmed that MHC disrupted by epigenetic can cause defective antigen processing and surface presentation therefore resistance to PD-1/PD-L1 blockade.
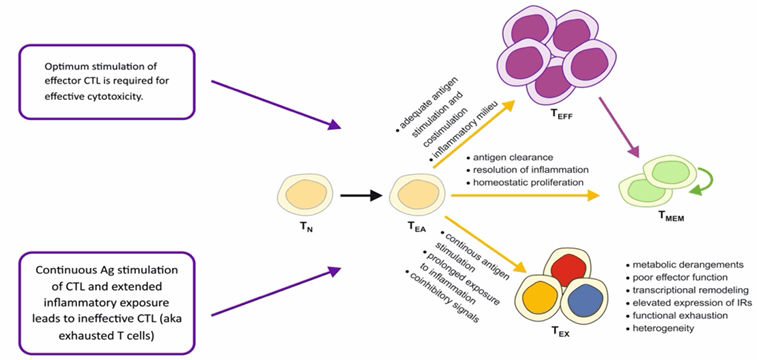
Figure 8: The process involved in generation of exhausted CD8+ T cells (TEX).
Another proposed mechanism of resistance is called T-cell exclusion, which means inhibition of CTL to infiltrate into TME, without the issue of defective antigen processing/presenting ability of APCs, by specific somatic mutation. Spranger et al. mouse model of melanoma showed an inverse relationship between increased activation of β-catenin/Wnt signalling, caused by β-catenin/Wnt mutation, and the number of CD4CD8 CTL in TME [79]. This phenomenon could explain as to why urothelial bladder cancer, a highly mutagenic tumor but suffers T-cell exclusion, is resistant to PD-1/PD-L1 blockade. Another culprit is mitogen activated protein kinase (MAPK) signalling which confers resistance PD-1/PD-L1 blockade, as this mutation leads to the release of anti T-cell recruitment cytokines such as vascular endothelial growth factor and interleukin 8 [80]. An immunosuppressive TME, as described above, is another contributor to primary resistance to PD-1/PD-L1 inhibition. Studies confirmed the role of intra-tumor T-regs and MDSCs in the resistance process [81, 82]. On another hand, acquired resistance could be due to ‘exhausted’ CD8+ T cells (TEX) (Figure 8) [83]. TEX is the result of persistent stimulation of PD-1 by tumor specific antigens. A study by Philip M et al. showed that at early stages the TEX state is reversible and provide a window of opportunity to PD-1 inhibition to work, however, a more prolonged exposure to tumor specific antigens causes irreversible epigenetic dysfunction that renders chromatin unreachable and impervious to more reconditioning and revival [84]. Another mechanism of acquired resistance to ICIs is loss of neoantigens.
Moreover, the higher the burden of mutations that is associated with a higher level of neoantigen within the cancer cells, the higher the likelihood of NSCLC responding to ICIs, loss of tumor neoantigen confers resistance to ICIs [85, 86]. Analysis of cancer cells from NSCLC tumors that initially responded to ICI then developed resistance hence tumor progression, showed genomic changes causing loss of 7 to 18 putative mutation-associated neoantigens within ICI resistant cancer cells. This acquired resistance is due to the cancer cells ability of eliminating both truncal and sub-clonal mutations [86]. The role of gut microbiome in predicting sensitivity or tolerance of cancer cells to ICIs, is the subject of several investigations [87]. For example, a study by Chaput et al. showed a favourable correlation between response to ICIs, in twenty-six patients with metastatic melanoma, and gut microbiota enriched with Faecalibacterium and other Firmicutes [88]. Conversely, Bacteroidales enriched gut microbiome is shown to be associated with resistance of cancer cells to ICIs [89]. Manipulating gut microbiome is therefore provide an opportunity to develop interventions to overcome resistance to ICIs. For example, a study of germ-free or antibiotic-treated mice receiving faecal microbiota transplantation (FMT) sourced from patients responded to ICIs, showed better response to treatment with ICIs, unlike mice receiving FMT from patients resisted treatment with ICIs [90].
Interestingly, there growing body of data showing that patients previously treated with ICIs then developed resistance to immunotherapy have become more suitable to the cytotoxic effect of chemotherapy [91]. This might be due to the supressing effect of immunotherapy on anti-cytotoxic pathways within TME or the cancer cells [69]. On the other hand, treatment with radiotherapy and chemotherapy could lead to further genomic instability in the cancer cells. Therefore, more mutation generating an array of new neo-epitopes which increases the tumor immunogenicity and its susceptibility to ICIs [85]. Oncolytic viruses injected to local but non-respectable melanoma are used in clinical settings as a mean to increase tumor immunogenicity abscopal effect [92]. The virus, talimogene laherparepvec, a genetically modified herpes simplex-1 virus expressing granulocyte-macrophage CSF licensed by FDA, causes tumor lysis which in turn releases neoantigens that stimulate anticancer T cells leading to reduction in the volume non-injected lesions [92].
The durable response of tumor to ICI is dependent on the quantity and the quality of memory T cells (TEM) infiltrating the tumor [93]. Paucity of or defective TEM infiltrated tumor result in the failure of this long-term response to ICI [94]. Tumors of high mutation burden, which is often associated with persistence of tumor antigen, are less amenable to repopulation with adequate quantity and quality of TEM [95]. Therefore, augmentation and clonal expansion of existing TEM or priming new T cells could overcome this mechanism of resistance to treatment with ICIs.
Conclusion and Future Directions
The discovery of immune checkpoint proteins and their inhibitors does represent a major breakthrough in the fight against cancer; it provided the potential for durable and, in some cases, curative response in notoriously aggressive cancers, such as non-small cell lung cancer (NSCLC) and melanoma. Although we witnessed disappointing results in some other cancers such as pancreatic and ureteral malignancies.
One of the future challenges is to study the effect of synergistically combining different immunotherapy modalities, such as cancer vaccines with ICI, without increasing the risk of autoimmunity. A phase 2 clinical trial of combining nivolumab with bevacizumab, an anti-angiogenic, showed encouraging results, in relapsed ovarian cancer [96]. Using such molecular based strategies, or combination of ICI with epigenetic therapy or conventional chemotherapy, to turn none immunogenic ‘cold’ to immunogenic ‘hot’ tumors should be studied further to elucidate mechanisms involved in the symbiosis of the mechanisms of action of different modalities of systemic therapies with the view of testing their safety and efficacy in randomized clinical trials. Using ICIs in neo-adjuvant setting to control micro-metastasis is another research area that requires more investigation. Translating the encouraging results, of mice model’s modification of gut microbiome to enhance tumor response to ICI, into human clinical trials is the focus of several research groups [72, 97].
In addition, there are preclinical trials underway testing the use of non-viral oncolytic interventions to induce abscopal effects in local but non-respectable tumors such as: microwave, locally injected cytotoxins, photodynamic therapy, electrochemotherapy, high-intensity focused ultrasonography, laser therapy and cryotherapy [98]. Moreover, there are ongoing studies investigating the efficacy of transgenic T cell receptor and chimeric antigen receptor (CAR) T cells in solid tumors. Genetically engineered (CAR) T cells have scFv domain, which has a high affinity to tumor-specific antigen, as well as a domain that triggers T-cell cytotoxicity [77].
HER2, EGFR and CEA that are overexpressed on the surface of cells in GI tumors and associated with metastasis, are targeted by bioengineered (CAR) T cells with some promising results [99]. The role of ICIs in the neoadjuvant setting is worth exploring in future research. Preliminary results of studies investigating treatment with ICI of respectable nodal positive NSCLC as part of multi-modality treatment, show encouraging results in term of side effect profile, not causing delay in surgery and evidence of pathological regression of the index tumor [100, 101]. The immunogenic effect of neoadjuvant immunotherapy on the primary tumor might lead to expansion and activation of tumor-specific T cells, which in turn promote antitumor surveillance and destruction of micro metastasis. The latter is thought to be a major cause of postsurgical recurrence of cancer. Finding expanded clones of T cells in tumor and peripheral blood of patients with early stage NSCLC that received neoadjuvant ICIs, support the hypothesis of better control of micro-metastasis via administering neoadjuvant ICIs [101].
Author Contributions
HS and KR conceived of the presented idea. HS developed the idea further and conducted review of literature. KR and SU verified the key findings and conclusion of the review. SZ supervised the design of the figures. RS reviewed the language aspect of the article and focused on improving the section related to resistance to immune checkpoint inhibitors. All authors discussed the results and contributed to the final manuscript.
Article Info
Article Type
Review ArticlePublication history
Received: Mon 24, Aug 2020Accepted: Fri 25, Sep 2020
Published: Wed 30, Sep 2020
Copyright
© 2023 Harman Saman. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited. Hosting by Science Repository.DOI: 10.31487/j.COR.2020.09.08
Figures & Tables
References
- Pennock GK, Chow LQ (2015) The Evolving Role of Immune Checkpoint Inhibitors in Cancer Treatment. Oncologist 20: 812-822. [Crossref]
- La Beck NM, Jean GW, Huynh C, Alzghari SK, Lowe DB (2015) Immune Checkpoint Inhibitors: New Insights and Current Place in Cancer Therapy. Pharmacotherapy 35: 963-976. [Crossref]
- Dunn GP, Bruce AT, Ikeda H, Old LJ, Schreiber RD (2002) Cancer immunoediting: from immunosurveillance to tumor escape. Nat Immunol 3: 991-998. [Crossref]
- Waldmann TA (2003) Immunotherapy: past, present and future. Nat Med 9: 269-277. [Crossref]
- Amin A, White RL Jr (2013) High-dose interleukin-2: is it still indicated for melanoma and RCC in an era of targeted therapies? Oncology (Williston Park) 27: 680-691. [Crossref]
- Finn OJ (2012) Immuno-oncology: understanding the function and dysfunction of the immune system in cancer. Ann Oncol 23: viii6- viii9. [Crossref]
- Diesendruck Y, Benhar I (2017) Novel immune check point inhibiting antibodies in cancer therapy-Opportunities and challenges. Drug Resist Updat 30: 39-47. [Crossref]
- Beatty GL, Gladney WL (2015) Immune escape mechanisms as a guide for cancer immunotherapy. Clin Cancer Res 21: 687-692. [Crossref]
- Martinez Lostao L, Anel A, Pardo J (2015) How Do Cytotoxic Lymphocytes Kill Cancer Cells? Clin Cancer Res 21: 5047-5056. [Crossref]
- Attarwala H (2010) TGN1412: From Discovery to Disaster. J Young Pharm 2: 332-336. [Crossref]
- O'Donnell JS, Teng MWL, Smyth MJ (2019) Cancer immunoediting and resistance to T cell-based immunotherapy. Nat Rev Clin Oncol 16: 151-67. [Crossref]
- Fulda S (2015) Promises and Challenges of Smac Mimetics as Cancer Therapeutics. Clin Cancer Res 21: 5030-5036. [Crossref]
- Voskoboinik I, Whisstock JC, Trapani JA (2015) Perforin and granzymes: function, dysfunction and human pathology. Nat Rev Immunol 15: 388-400. [Crossref]
- Bossi G, Griffiths GM (2005) CTL secretory lysosomes: biogenesis and secretion of a harmful organelle. Semin Immunol 17: 87-94. [Crossref]
- Smyth MJ, Thia KY, Street SE, MacGregor D, Godfrey DI et al. (2000) Perforin-mediated cytotoxicity is critical for surveillance of spontaneous lymphoma. J Exp Med 192: 755-760. [Crossref]
- Macagno M, Bandini S, Stramucci L, Quaglino E, Conti L et al. (2014) Multiple roles of perforin in hampering ERBB-2 (Her-2/neu) carcinogenesis in transgenic male mice. J Immunol 192: 5434-5441. [Crossref]
- Coulie PG, Van den Eynde BJ, van der Bruggen P, Boon T (2014) Tumour antigens recognized by T lymphocytes: at the core of cancer immunotherapy. Nat Rev Cancer 14: 135-146. [Crossref]
- Tran E, Turcotte S, Gros A, Robbins PF, Lu Y et al. (2014) Cancer immunotherapy based on mutation-specific CD4+ T cells in a patient with epithelial cancer. Science 344: 641-645. [Crossref]
- Topalian SL, Hodi FS, Brahmer JR, Gettinger SN, Smith DC et al. (2012) Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med 366: 2443-2454. [Crossref]
- Campoli M, Ferrone S (2008) HLA antigen changes in malignant cells: epigenetic mechanisms and biologic significance. Oncogene 27: 5869-5885. [Crossref]
- Taube JM, Anders RA, Young GD, Xu H, Sharma R et al. (2012) Colocalization of inflammatory response with B7-h1 expression in human melanocytic lesions supports an adaptive resistance mechanism of immune escape. Sci Transl Med 4: 127-137. [Crossref]
- Coussens LM, Zitvogel L, Palucka AK (2013) Neutralizing tumor-promoting chronic inflammation: a magic bullet? Science 339: 286-291. [Crossref]
- Liu M, Zhou J, Chen Z, Cheng AS (2017) Understanding the epigenetic regulation of tumours and their microenvironments: opportunities and problems for epigenetic therapy. J Pathol 241: 10-24. [Crossref]
- Talmadge JE, Gabrilovich DI (2013) History of myeloid-derived suppressor cells. Nat Rev Cancer 13: 739-752. [Crossref]
- Ostrand Rosenberg S, Sinha P, Beury DW, Clements VK (2012) Cross-talk between myeloid-derived suppressor cells (MDSC), macrophages, and dendritic cells enhances tumor-induced immune suppression. Semin Cancer Biol 22: 275-281. [Crossref]
- Pan PY, Ma G, Weber KJ, Ozao Choy J, Wang G et al. (2010) Immune stimulatory receptor CD40 is required for T-cell suppression and T regulatory cell activation mediated by myeloid-derived suppressor cells in cancer. Cancer Res 70: 99-108. [Crossref]
- Ugel S, De Sanctis F, Mandruzzato S, Bronte V (2015) Tumor-induced myeloid deviation: when myeloid-derived suppressor cells meet tumor-associated macrophages. J Clin Invest 125: 3365-3376. [Crossref]
- Geissmann F, Manz MG, Jung S, Sieweke MH, Merad M et al. (2010) Development of monocytes, macrophages, and dendritic cells. Science 327: 656-661. [Crossref]
- Strauss L, Sangaletti S, Consonni FM, Szebeni G, Morlacchi S et al. (2015) RORC1 Regulates Tumor-Promoting "Emergency" Granulo-Monocytopoiesis. Cancer Cell 28: 253-269. [Crossref]
- Tan W, Zhang W, Strasner A, Grivennikov S, Cheng JQ et al. (2011) Tumour-infiltrating regulatory T cells stimulate mammary cancer metastasis through RANKL-RANK signalling. Nature 470: 548-553. [Crossref]
- Kalluri R (2016) The biology and function of fibroblasts in cancer. Nat Rev Cancer 16: 582-598. [Crossref]
- Sahai E, Astsaturov I, Cukierman E, DeNardo DG, Egeblad M et al. (2020) A framework for advancing our understanding of cancer-associated fibroblasts. Nat Rev Cancer 20:174-186. [Crossref]
- Liang X, Liu Y, Mei S, Zhang M, Xin J et al. (2015) MicroRNA-22 impairs anti-tumor ability of dendritic cells by targeting p38. PLoS One 10: e0121510. [Crossref]
- Lee PP, Fitzpatrick DR, Beard C, Jessup HK, Lehar S et al. (2001) A critical role for Dnmt1 and DNA methylation in T cell development, function, and survival. Immunity 15: 763-774. [Crossref]
- Pardoll DM (2012) The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer 12: 252-264. [Crossref]
- Schadendorf D, Hodi FS, Robert C, Weber JS, Margolin K et al. (2015) Pooled Analysis of Long-Term Survival Data From Phase II and Phase III Trials of Ipilimumab in Unresectable or Metastatic Melanoma. J Clin Oncol 33: 1889-1894. [Crossref]
- Ribas A, Hamid O, Daud A, Hodi FS, Wolchok JD et al. (2016) Association of Pembrolizumab With Tumor Response and Survival Among Patients With Advanced Melanoma. JAMA 315: 1600-1609. [Crossref]
- Larkin J, Hodi FS, Wolchok JD (2015) Combined Nivolumab and Ipilimumab or Monotherapy in Untreated Melanoma. N Engl J Med 373: 1270-1271. [Crossref]
- Elias R, Giobbie Hurder A, McCleary NJ, Ott P, Hodi FS et al. (2018) Efficacy of PD-1 & PD-L1 inhibitors in older adults: a meta-analysis. J Immunother Cancer 6: 26. [Crossref]
- Zhang L, Sun L, Yu J, Shan F, Zhang K et al. (2019) Comparison of Immune Checkpoint Inhibitors between Older and Younger Patients with Advanced or Metastatic Lung Cancer: A Systematic Review and Meta-Analysis. Biomed Res Int 2019: 9853701. [Crossref]
- Ninomiya K, Oze I, Kato Y, Kubo T, Ichihara E et al. (2020) Influence of age on the efficacy of immune checkpoint inhibitors in advanced cancers: a systematic review and meta-analysis. Acta Oncol 59: 249-256. [Crossref]
- Wallis CJD, Butaney M, Satkunasivam R, Freedland SJ, Patel SP et al. (2019) Association of Patient Sex With Efficacy of Immune Checkpoint Inhibitors and Overall Survival in Advanced Cancers: A Systematic Review and Meta-analysis. JAMA Oncol 5: 529-536. [Crossref]
- Myers G (2018) Immune-related adverse events of immune checkpoint inhibitors: a brief review. Curr Oncol 25: 342-37. [Crossref]
- Postow MA, Sidlow R, Hellmann MD (2018) Immune-Related Adverse Events Associated with Immune Checkpoint Blockade. N Engl J Med 378: 158-168. [Crossref]
- Weinmann SC, Pisetsky DS (2019) Mechanisms of immune-related adverse events during the treatment of cancer with immune checkpoint inhibitors. Rheumatology (Oxford) 58: vii59-vii67. [Crossref]
- Day D, Hansen AR (2016) Immune-Related Adverse Events Associated with Immune Checkpoint Inhibitors. BioDrugs 30: 571-584. [Crossref]
- Waterhouse P, Penninger JM, Timms E, Wakeham A, Shahinian A et al. (1995) Lymphoproliferative disorders with early lethality in mice deficient in Ctla-4. Science 270: 985-988. [Crossref]
- Granier C, De Guillebon E, Blanc C, Roussel H, Badoual C et al. (2017) Mechanisms of action and rationale for the use of checkpoint inhibitors in cancer. ESMO Open 2: e000213. [Crossref]
- Nishimura H, Okazaki T, Tanaka Y, Nakatani K, Hara M et al. (2001) Autoimmune dilated cardiomyopathy in PD-1 receptor-deficient mice. Science 291: 319-322. [Crossref]
- Zhou X, Yao Z, Yang H, Liang N, Zhang X et al. (2020) Are immune-related adverse events associated with the efficacy of immune checkpoint inhibitors in patients with cancer? A systematic review and meta-analysis. BMC Med 18: 87. [Crossref]
- Tison A, Quere G, Misery L, Funck Brentano E, Danlos FX et al. (2019) Safety and Efficacy of Immune Checkpoint Inhibitors in Patients With Cancer and Preexisting Autoimmune Disease: A Nationwide, Multicenter Cohort Study. Arthritis Rheumatol 71: 2100-2111. [Crossref]
- Sharma P, Hu Lieskovan S, Wargo JA, Ribas A (2017) Primary, Adaptive, and Acquired Resistance to Cancer Immunotherapy. Cell 168 : 707-723. [Crossref]
- Magrone T, Jirillo E (2018) Update on Mechanisms of Adaptive Resistance to Immune Check Point Blockers in Malignancies: A Short Commentary. Curr Pharm Des 24: 5349-5351. [Crossref]
- Ribas A, Shin DS, Zaretsky J, Frederiksen J, Cornish A et al. (2016) PD-1 Blockade Expands Intratumoral Memory T Cells. Cancer Immunol Res 4: 194-203. [Crossref]
- Yearley JH, Gibson C, Yu N, Moon C, Murphy E et al. (2017) PD-L2 Expression in Human Tumors: Relevance to Anti-PD-1 Therapy in Cancer. Clin Cancer Res 23: 3158-3167. [Crossref]
- Brown JA, Dorfman DM, Ma FR, Sullivan EL, Munoz O et al. (2003) Blockade of programmed death-1 ligands on dendritic cells enhances T cell activation and cytokine production. J Immunol 170: 1257-1266. [Crossref]
- Wang C, Thudium KB, Han M, Wang XT, Huang H et al. (2014) In vitro characterization of the anti-PD-1 antibody nivolumab, BMS-936558, and in vivo toxicology in non-human primates. Cancer Immunol Res 2: 846-856. [Crossref]
- Ibrahim R, Stewart R, Shalabi A (2015) PD-L1 blockade for cancer treatment: MEDI4736. Semin Oncol 42: 474-483. [Crossref]
- Centanni M, Moes D, Troconiz IF, Ciccolini J, van Hasselt JGC (2019) Clinical Pharmacokinetics and Pharmacodynamics of Immune Checkpoint Inhibitors. Clin Pharmacokinet 58: 835-857. [Crossref]
- Wang W, Wang EQ, Balthasar JP (2008) Monoclonal antibody pharmacokinetics and pharmacodynamics. Clin Pharmacol Ther 84: 548-558. [Crossref]
- Darvin P, Toor SM, Sasidharan Nair V, Elkord E (2018) Immune checkpoint inhibitors: recent progress and potential biomarkers. Exp Mol Med 50: 1-11. [Crossref]
- Curry WT, Lim M (2015) Immunomodulation: checkpoint blockade etc. Neuro Oncol 17: vii26-vii31. [Crossref]
- Taube JM, Klein A, Brahmer JR, Xu H, Pan X et al. (2014) Association of PD-1, PD-1 ligands, and other features of the tumor immune microenvironment with response to anti-PD-1 therapy. Clin Cancer Res 20: 5064-5074. [Crossref]
- Wolchok JD, Neyns B, Linette G, Negrier S, Lutzky J et al. (2010) Ipilimumab monotherapy in patients with pretreated advanced melanoma: a randomised, double-blind, multicentre, phase 2, dose-ranging study. Lancet Oncol 11: 155-164. [Crossref]
- Rizvi NA, Hellmann MD, Snyder A, Kvistborg P, Makarov V et al. (2015) Cancer immunology. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science 348: 124-128. [Crossref]
- Lipson EJ, Velculescu VE, Pritchard TS, Sausen M, Pardoll DM et al. (2014) Circulating tumor DNA analysis as a real-time method for monitoring tumor burden in melanoma patients undergoing treatment with immune checkpoint blockade. J Immunother Cancer 2: 42. [Crossref]
- Twyman Saint Victor C, Rech AJ, Maity A, Rengan R, Pauken KE et al. (2015) Radiation and dual checkpoint blockade activate non-redundant immune mechanisms in cancer. Nature 520: 373-377. [Crossref]
- Hamilton G, Rath B (2017) Avelumab: combining immune checkpoint inhibition and antibody-dependent cytotoxicity. Expert Opin Biol Ther 17: 515-523. [Crossref]
- Lee JY, Lee HT, Shin W, Chae J, Choi J et al. (2016) Structural basis of checkpoint blockade by monoclonal antibodies in cancer immunotherapy. Nat Commun 7: 13354. [Crossref]
- Liu K, Tan S, Chai Y, Chen D, Song H et al. (2017) Structural basis of anti-PD-L1 monoclonal antibody avelumab for tumor therapy. Cell Res 27: 151-153. [Crossref]
- Tan S, Liu K, Chai Y, Zhang CW, Gao S et al. (2018) Distinct PD-L1 binding characteristics of therapeutic monoclonal antibody durvalumab. Protein Cell 9: 135-139. [Crossref]
- Rodallec A, Sicard G, Fanciullino R, Benzekry S, Lacarelle B et al. (2018) Turning cold tumors into hot tumors: harnessing the potential of tumor immunity using nanoparticles. Expert Opin Drug Metab Toxicol 14: 1139-1147. [Crossref]
- Gubin MM, Zhang X, Schuster H, Caron E, Ward JP et al. (2014) Checkpoint blockade cancer immunotherapy targets tumour-specific mutant antigens. Nature 515: 577-581. [Crossref]
- Schumacher TN, Schreiber RD (2015) Neoantigens in cancer immunotherapy. Science 348: 69-74. [Crossref]
- Martin AM, Nirschl TR, Nirschl CJ, Francica BJ, Kochel CM et al. (2015) Paucity of PD-L1 expression in prostate cancer: innate and adaptive immune resistance. Prostate Cancer Prostatic Dis 18: 325-332. [Crossref]
- Chodon T, Comin Anduix B, Chmielowski B, Koya RC, Wu Z et al. (2014) Adoptive transfer of MART-1 T-cell receptor transgenic lymphocytes and dendritic cell vaccination in patients with metastatic melanoma. Clin Cancer Res 20: 2457-2465. [Crossref]
- Robbins PF, Morgan RA, Feldman SA, Yang JC, Sherry RM et al. (2011) Tumor regression in patients with metastatic synovial cell sarcoma and melanoma using genetically engineered lymphocytes reactive with NY-ESO-1. J Clin Oncol 29: 917-924. [Crossref]
- Vo DD, Prins RM, Begley JL, Donahue TR, Morris LF et al. (2009) Enhanced antitumor activity induced by adoptive T-cell transfer and adjunctive use of the histone deacetylase inhibitor LAQ824. Cancer Res 69: 8693-8699. [Crossref]
- Spranger S, Bao R, Gajewski TF (2015) Melanoma-intrinsic beta-catenin signalling prevents anti-tumour immunity. Nature 523: 231-235. [Crossref]
- Loi S, Dushyanthen S, Beavis PA, Salgado R, Denkert C et al. (2016) RAS/MAPK Activation Is Associated with Reduced Tumor-Infiltrating Lymphocytes in Triple-Negative Breast Cancer: Therapeutic Cooperation Between MEK and PD-1/PD-L1 Immune Checkpoint Inhibitors. Clin Cancer Res 22: 1499-1509. [Crossref]
- Ostrand Rosenberg S (2010) Myeloid-derived suppressor cells: more mechanisms for inhibiting antitumor immunity. Cancer Immunol Immunother 59: 1593-1600. [Crossref]
- Arce Vargas F, Furness AJS, Solomon I, Joshi K, Mekkaoui L et al. (2017) Fc-Optimized Anti-CD25 Depletes Tumor-Infiltrating Regulatory T Cells and Synergizes with PD-1 Blockade to Eradicate Established Tumors. Immunity 46: 577-586. [Crossref]
- Wherry EJ, Kurachi M (2015) Molecular and cellular insights into T cell exhaustion. Nat Rev Immunol 15: 486-499. [Crossref]
- Philip M, Fairchild L, Sun L, Horste EL, Camara S et al. (2017) Chromatin states define tumour-specific T cell dysfunction and reprogramming. Nature 545: 452-456. [Crossref]
- McGranahan N, Furness AJ, Rosenthal R, Ramskov S, Lyngaa R et al. (2016) Clonal neoantigens elicit T cell immunoreactivity and sensitivity to immune checkpoint blockade. Science 351: 1463-1469. [Crossref]
- Anagnostou V, Smith KN, Forde PM, Niknafs N, Bhattacharya R et al. (2017) Evolution of Neoantigen Landscape during Immune Checkpoint Blockade in Non-Small Cell Lung Cancer. Cancer Discov 7: 264-276. [Crossref]
- Esfahani K, Roudaia L, Buhlaiga N, Del Rincon SV, Papneja N et al. (2020) A review of cancer immunotherapy: from the past, to the present, to the future. Curr Oncol 27: S87-S97. [Crossref]
- Chaput N, Lepage P, Coutzac C, Soularue E, Le Roux K et al. (2017) Baseline gut microbiota predicts clinical response and colitis in metastatic melanoma patients treated with ipilimumab. Ann Oncol 28: 1368-1379. [Crossref]
- Gopalakrishnan V, Spencer CN, Nezi L, Reuben A, Andrews MC et al. (2018) Gut microbiome modulates response to anti-PD-1 immunotherapy in melanoma patients. Science 359: 97-103. [Crossref]
- Routy B, Le Chatelier E, Derosa L, Duong CPM, Alou MT et al. (2018) Gut microbiome influences efficacy of PD-1-based immunotherapy against epithelial tumors. Science 359: 91-97. [Crossref]
- Dwary AD, Master S, Patel A, Cole C, Mansour R et al. (2017) Excellent response to chemotherapy post immunotherapy. Oncotarget 8: 91795-91802. [Crossref]
- Harrington K, Freeman DJ, Kelly B, Harper J, Soria JC (2019) Optimizing oncolytic virotherapy in cancer treatment. Nat Rev Drug Discov 18: 689-706. [Crossref]
- Jenkins RW, Barbie DA, Flaherty KT (2018) Mechanisms of resistance to immune checkpoint inhibitors. Br J Cancer 118: 9-16. [Crossref]
- O'Donnell JS, Long GV, Scolyer RA, Teng MW, Smyth MJ (2017) Resistance to PD1/PDL1 checkpoint inhibition. Cancer Treat Rev 52: 71-81. [Crossref]
- Benci JL, Xu B, Qiu Y, Wu TJ, Dada H et al. (2016) Tumor Interferon Signaling Regulates a Multigenic Resistance Program to Immune Checkpoint Blockade. Cell 167: 1540e12-1554e12. [Crossref]
- Liu JF, Herold C, Gray KP, Penson RT, Horowitz N et al. (2019) Assessment of Combined Nivolumab and Bevacizumab in Relapsed Ovarian Cancer: A Phase 2 Clinical Trial. JAMA Oncol 5: 1731-1738. [Crossref]
- Yi M, Yu S, Qin S, Liu Q, Xu H et al. (2018) Gut microbiome modulates efficacy of immune checkpoint inhibitors. J Hematol Oncol 11: 47. [Crossref]
- Kepp O, Marabelle A, Zitvogel L, Kroemer G (2020) Oncolysis without viruses-inducing systemic anticancer immune responses with local therapies. Nat Rev Clin Oncol 17: 49-64. [Crossref]
- Zhang Q, Zhang Z, Peng M, Fu S, Xue Z et al. (2016) CAR-T cell therapy in gastrointestinal tumors and hepatic carcinoma: From bench to bedside. Oncoimmunology 5: e1251539. [Crossref]
- Wu Z, Man S, Sun R, Li Z, Wu Y et al. (2020) Recent advances and challenges of immune checkpoint inhibitors in immunotherapy of non-small cell lung cancer. Int Immunopharmacol 85: 106613. [Crossref]
- Forde PM, Chaft JE, Smith KN, Anagnostou V, Cottrell TR et al. (2018) Neoadjuvant PD-1 Blockade in Resectable Lung Cancer. N Engl J Med 378: 1976-1986. [Crossref]