Rett Syndrome: Search for Prognostic Factors
A B S T R A C T
Rett (RTT) syndrome is primarily a disorder of synaptic plasticity. The maturation of neurons is blocked resulting arrest of brain growth. Clinical signs of excitatory activity occur in early age but reduced in later childhood. Correcting this imbalance may be reflected in molecular changes. Our aim was to find prognostic factors using combination of clinical examination, neuroradiological and biochemical analysis of cerebrospinal fluid (CSF). The evaluation of 15 children with RTT included careful clinical examination, measurement of serum- and CSF-insulin-like growth factor-1 and -nerve growth factor, CSF-glutamate, -adrenocorticotropin (ACTH) and -acetyltransferase, genetic analysis, and single photon computed tomography (SPECT) studies. Predictive data for poor outcome of six children were early onset of clinical signs, early deceleration of head growth, and hypometabolism in SPECT images corresponding low CSF-NGF found in patients with RTT. These signs correlated with severity of later gross motor signs (no gait). Favorable outcomes were seen in nine children with late onset of the clinical symptoms and late deceleration of head growth. Two of these patients had the highest CSF-acetylcholinesterase and mutation R294X, and five had R306C mutation. ACTH/glutamate-ratio was significantly higher in the early onset-group compared to the late onset-group. This might mean an attempt to restore the balance between excitatory and inhibitory transmission. In conclusion, the clinical signs and hypo-perfusion in SPECT were the most predictive outcome measures. Our findings might be valuable because they impact the understanding of mechanisms of RTT and the potential to discovery of biological markers for proper diagnosis and treatment.
Keywords
Rett syndrome, predictive factors, head growth, SPECT, molecular factors
Introduction
Rett syndrome (RTT) is a neurodevelopmental disorder caused by mutations in MECP2 gene and first described fifty years ago. However, few predictive factors are known for the outcome of RTT. There are age-related clinical changes in RTT including head growth deceleration, loss of locomotor activity and shift in the balance between excitation to inhibition in electrical activity. It has been suggested that neurotransmitters and neurotrophins are involved in the pathogenic mechanisms of RTT. Advances in understanding of synaptic abnormalities were reported recently [1]. Extensive studies in a small population of RTT have been done in the Children`s Hospital, University of Helsinki, Finland to find pathogenic mechanisms for RTT [2]. Herein, we aimed to find clinical and molecular data for the predictive outcome of patients in context of new understanding of neuropathological bases of RTT [1].
In the Finnish studies, cerebrospinal fluid (CSF) glutamate was high [3]. The following four neurotrophic factors were measured: nerve growth factor (NGF), brain-derived neurotrophic factor (BDNF), glial cell line-derived neurotrophic factor (GDNF), and insulin-like growth factor 1 (IGF-1) from the frozen CSF and from serum by enzyme-linked immunosorbent assay (ELISA) [4]. The CSF-NGF was low compared to age-matched controls [5]. Levels of CSF-BDNF and -GDNF were below the limit of sensitivity of the methods used [6]. Neither CSF-IGF, nor serum-GDNF and -BDNF levels differ from the controls [6]. As far as we know, no CSF neurotrophic factors, CSF-adrenocorticotropin (ACTH) or -acetylcholinesterase studies, except ours, have been done earlier in RTT [7]. In the present study combined earlier and new data has been evaluated with a new aspect whether they could be of prognostic importance. There are only a few predictive factors for the outcome of RTT. Here we present some predictive outcome measures for RTT.
Material and Methods
Fifteen children with classic RTT were admitted to the Children`s Hospital, University of Helsinki and carefully evaluated for pathogenic mechanisms [8]. Their ages ranged between 1.8 and 17.4 years (median 4.2). The patients were studied at specified limited time period. We evaluated now our data of these children in two parameters: i. early/late onset of the symptoms, ii. ages before/after 8 years.
i) Early/late onset of the symptoms: clinical normality and deceleration of the head growth : Patients with early onset of the symptoms, patients 1-6 (Table 1) had normal development only up to 5 or 8 months of age and regression thereafter. Patients with late onset of the symptoms, patients 7-15 had had normal development longer than 10 (median 15, maximum 72) months of age and regression later.
ii) Age before/after 8 years, because of bi-phasic changes in the clinical conditions of girls with RTT concerning abnormalities in glutamate excitatory synapses reported in several studies, we have evaluated the biochemical studies in these two age groups [9-12].
All patients had careful clinical examinations including growth parameters, hyperventilation, EEG and neuroradiological examinations (CT/MRI). Nine of the 15 had a single photon emission tomography (SPECT) study. All patients had a number of biochemical investigations but not all investigations could be done in all patients (See below). Controls for the biochemical studies (n=68) were age-matched (aged 0.5-17 years) children with neurological symptoms. It would have been unethical to perform lumbar puncture on healthy children. In all of whom CSF was taken as a part of diagnostic examinations, mostly for exclusion of infection of the central nervous system (CNS), no infections were found. We performed a careful search for every patient suitable to be a control. We excluded subjects who had factors that could have affected the results, e.g. those having progressive disease, more severe than mild symptoms (glutamate), malnutrition, diabetes, hypothyroidism, hepatic failure, chronic inflammation (IGF-1), epilepsy and mental retardation (NGF). We also had 15 age-matched healthy controls from surgery patients (IGF-1 and BDNF).
Head circumference was plotted on the Finnish normal growth chart and expressed in in standard deviation (SD) from mean in healthy population. The measurements were made at each (frequent) visit to Department of Child Neurology, Hospital for Children and Adolescents, University of Helsinki. Brain perfusion was studied with the 99m Tc-hexamethyl-propylenamine oxime method (Amersham International plc, Amersham, UK). SPECT examinations were performed with a Picker International DDC 4096 single-detector rotating camera (Cleveland, OH, USA) equipped with a low-energy general-purpose parallel hole collimator and NUD SPETS Software (Nuclear Diagnostics, Stockholm, Sweden). Data were processed with Butterworth filter and a ramp filter used for image reconstruction. Brain perfusion SPECT images were evaluated visually and using region of interest (ROI) calculations. Semi-quantitative analysis based on regional side-to-side differences and cerebrum-to-cerebellum ratios were calculated from the stored material (Figure 1). The hypoperfusion findings were graded as mild, moderate and severe [13]. The two nuclear medicine specialists on our team did not have access to clinical data when evaluating the SPECT scans. Because of ethical constraints SPECT data from age-matched healthy children could not be collected for comparison with data of the patients.

Figure 1: ROI map used in semiqantitation of brain perfusion studies.
All the CSF samples of RTT patients were frozen immediately in liquid nitrogen and stored at –70oC until analysis. The determinations of CSF-ACTH, -acetyl cholinesterase, -glutamate and -IGF-1 were all made at Laboratory of Helsinki, University of Helsinki. CSF-ACTH was assayed by the automated immune-analyzer Immulite 2000 (Siemens, LIanberis, Caernarfon, United Kingdom) by a commercial radioimmunoassay. Samples of the patients and controls we collected between 08:00 and 13:00 hours to avoid false interpretation due to circadian rhythm. CSF-acetyl cholinesterase activity was measured using colorimetric methods described in detail earlier [14]. CSF-glutamate was studied by reverse-phase high-performance liquid chromatography and precolum α-phtaldialdehyde derivatization method as described [15]. CSF-IGF-1 was determined by means of radioimmunoassay using a commercially kit (Mediagnost, Tuebingen, Germany) according to the instructions of the manufacturer. The samples were analyzed in duplicate. The inter-assay coefficient of variations was less than 10%. The sensitivity of the assay was 0.02 µg/l.
Serum- and CSF-NGF were determined with a sensitive (two-site) ELISA as described in detail earlier by Lindholm et al. and Lappalainen et al. [5, 16]. The determination was made in Department of Neurochemistry, Max-Planck Institute for Psychiatry, Martinsried, Germany. The human NGF used as the standard was a gift from Genentech, San Francisco, CA, USA. A monoclonal anti-NGF-antibody (Boehring, Mannheim, Germany) was used. A genetic study was performed for 13 of the 15 patients. Mutation types of the Finnish patients with RTT were analyzed in a separate study [17]. PCR amplification of the coding exons of MECP2 was performed as described elsewhere with minor modifications [18]. The study was made at the Department of Human Molecular Genetics, University of Helsinki.
I Epilepsy
Ten of 15 patients had epilepsy at the time of the evaluation. The age of onset of epilepsy and antiepileptic treatment is seen in (Table 1).
II Outcome Measures
We used the ability to walk as the main outcome measure for gross motor function: with the result of gait/ no gait.
III Statistics
The data were analyzed with the IBM SPSS Statistics for Windows, Version 22.0. (IBM Corp., Armonk, NY, USA). The data are presented as the number of cases, mean (SD) or median (minimum-maximum), as appropriate. The statistical analyses were performed with the Mann Whitney U-test. A p-value of 0.05 was considered as the limit of statistical significance.
Table 1: Patients’ characteristics. Comparison of early onset-group to late onset-group, p-values Mann-Whitney-test.
|
|
Normal mo |
HC (SD) |
Epilepsy yrs:mo |
AED |
CSF ACTH |
CSF Glutam |
CSF IGF-1 |
CSF Acetyl |
CSF NGF |
Gene |
Gait mo |
SPECT |
|||
|
yrs:mo |
Front |
Temp |
Par |
||||||||||||
|
Early onset 6-8 months, N=6 |
|||||||||||||||
|
#1 / 1.7 yrs |
6 |
-2 |
No |
No |
43 |
265 |
0.53 |
97.7 |
0.1 |
R255X |
No |
|
R++/ L+++ |
R+ |
|
|
#2 / 4.2 yrs. |
6 |
-3.5 |
No |
No |
ND |
407 |
0.56 |
ND |
6.6 |
R270X |
No |
4:2 |
R+++/ L++ |
T++ |
R++/ L+ |
|
#3 / 4.2yrs |
6 |
-2.5 |
1:8 |
VPA |
25 |
213 |
0.16 |
72.5 |
0.16 |
G269X |
No |
ND |
|
|
|
|
#4 / 4.2 yrs |
6 |
ND |
1:6 |
VPA |
|
|
|
ND |
|
|
No |
4:2 |
R+++/L++ |
TR+++ |
|
|
#5 / 17.4 yrs |
8 |
-3.0 |
1:7 |
VPA |
36 |
252 |
0.44 |
56.0 |
0.1 |
ND |
No |
17:5 |
R+/L++ |
|
L+ |
|
#6 /11.4 yrs |
5 |
-3.0 |
4:4 |
VPA |
34 |
436 |
0.61 |
55.1 |
0.1 |
R302A |
20 / later lost |
11:6 |
Hypometabolism |
||
|
Late onset 10-24 months, N=9 |
|||||||||||||||
|
#7 / 16.5 yrs |
12 |
-0.8 |
2:6 |
CBZ/VPA |
26 |
332 |
0.29 |
ND |
0.4 |
R306C |
18 |
ND |
|
|
|
|
#8 / 11.3yrs |
10 |
-1.5 |
4:0 |
CBZ |
ND |
ND |
0.74 |
66.2 |
0.1 |
R306C |
21 |
11:4 |
Normal |
||
|
#9 / 2.9 yrs |
15 |
-2 |
No |
No |
31 |
521 |
0.47 |
60.3 |
0.1 |
R106W |
14 |
3:2 |
|
R+ |
|
|
#10 / 3yrs.11mo |
15 |
-0.3 |
1:6 |
VPA |
40 |
539 |
0.48 |
53.9 |
ND |
R306C |
20 |
3:5 |
R++/L++ |
|
L+ |
|
#11 /6.5 yrs. |
23 |
-1.5 |
4:0 |
VPA |
ND |
361 |
ND |
ND |
6.7 |
R306C |
20 |
ND |
|
|
|
|
#12 / 9.8 yrs |
18 |
-1.7 |
3:7 |
CBZ |
23 |
311 |
0.48 |
61.4 |
3.0 |
R306C |
16 |
9:10 |
R+ |
|
R+ |
|
#13 / 2.3 yrs |
72 |
-1.3 |
No |
No |
ND |
ND |
0.16 |
135.8 |
2.7 |
R294X |
12 |
2:7 |
R+ |
|
|
|
#14 / 13.4 yrs |
18 |
-0.8 |
2:0 |
CBZ / VPA |
ND |
270 |
0.79 |
127.4 |
0.1 |
R294X |
16 |
13:5 |
|
R+/L+ |
|
|
#15 / 2 yrs |
ND |
0 |
No |
No |
29 |
|
|
66.3 |
|
T158M |
20 |
ND |
|
|
|
|
P-value |
|||||||||||||||
|
early vs. late |
|
0.002 |
|
|
0.41 |
0.25 |
1.0 |
0.51 |
0.53 |
|
|
|
|
|
|
Abbreviations: mo=months, yrs=years, HC: head circumference, AED: antiepileptic drugs, ACTH: adrenocorticotrophic hormone, CSF: cerebrospinal fluid, IGF-1: insulin-like growth factor-1, Glutam: glutamate, Acetyl: acetylcholine esterase; NGF: beta nerve growth factor, II-IV: Stages of Rett syndrome; ND: :no data, Front: frontal lobe; Temp: temporal lobe; Par: parietal lobe; R:right; L:left, + = slight; ++ = moderate, +++ = severe; N: normal.
Results
Table 1 summarizes the clinical and laboratory results of the patients.
I Head Circumference
The mean head circumference of the early onset patients was significantly smaller than in patients with late onset. Patients with early onset of symptoms had mean head circumference that were -2.8 SD below normal (minimum -3.5, maximum -2), while patients with late onset only showed a decrease of -1.1 SD (-1.7, 0) (p=0.002). The head growth deceleration was noticed earlier (between 4 and 8 months) in the early onset-group than in the late onset-group (at 2-3 years of age). The head growth deceleration was reported when there was the first slowing-down from the normal range (Table 1).
II Hypoperfusion in the SPECT Study
All five patients with early onset symptoms studied had hypo-perfusion on SPECT. This was severe in three and moderate in one. The grade of hypoperfusion was not given in the fifth. The site of hypoperfusion was predominantly frontal (Table 1). Of the six patients with late onset, only one (Patient 10 with onset of epilepsy at 18 months) had moderate hypoperfusion in the frontal lobes; the other patients had only minimal hypoperfusion (n=4) or none (n=1).
III Biochemical Results
The biochemical results are shown in (Table 2), early onset-/late onset-groups, and (Table 3), age below /above 8 years.
IV CSF levels of ACTH, Glutamate, NGF and IGF-1
Overall mean CSF-ACTH levels were higher in patients with RTT (33. 8 ng/ml (SD 6.6) than in controls (24.6 ng/ml (SD 5.9); (Figure 2A), p= 0.009). CSF levels of glutamate also were higher in patients with RTT (355 nmol/l (SD 109)) compared to controls (204 nmol/l (SD 55.5); (Figure 2B), p= 0.0006). The ACTH/glutamate-ratio was significantly higher in the early onset-group (mean 0.11 (SD 0.032) compared to late onset group (0.07 (SD 0.01); p= 0.032). In contrast, CSF levels of NGF were significantly lower in patients with RTT (1.7 pg/ml (SD 2.5)) versus controls (7.0 pg/ml (SD 5.7); (Figure 2C), (p=0.001). Levels of CSF-IGF-1 did not differ between controls (0.46 ug/l (SD 0.20)) and patient with RTT (0.56 ug/l (SD 0.23), (Figure 2D), p=0.22) nor were there differences in CSF-IGF-1 in younger versus older patients (p=0.24; Table 3).
Table 2: Comparisons of concentrations of biochemical parameters in patients with an early (regression of development before age of 5-8 months) versus late onset (regression of development at age of 10-24 months) of RTT. Data are mean (SD).
|
Parameter |
Early onset n=6 |
Late onset n=9 |
P-value |
|
CSF-ACTH ng/ml |
34.5 (7.4) |
29.8 (6.5) |
0.41 |
|
CSF-glutamate nmol/l |
315 (100) |
389 (113) |
0.25 |
|
CSF-IGF-1 µg/l |
0.46 (0.18) |
0.49 (0.22) |
1.0 |
|
CSF-ACTH/glutamate-ratio |
0.11 (0.03) |
0.07 (0.01) |
0.032 |
|
CSF-IGF-1/glutamate-ratio |
0.0014 (0.0005) |
0.0014 (0.0009) |
0.84 |
|
CSF-Acetylcholinesterase activity nmol/mg prot/minute |
70.3 (19.9) |
81.6 (34.5) |
0.79 |
|
CSF-Nerve growth factor pg/ml |
2.27 (3.75) |
1.87 (2.48) |
0.53 |
CSF=cerebrospinal fluid, ACTH=adrenocorticotropic hormone, IGF= insulin-like growth factor.
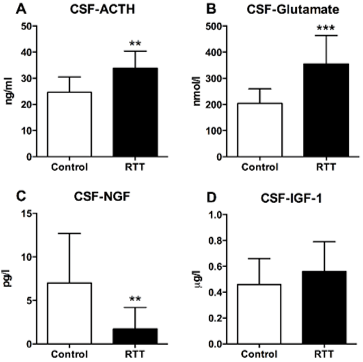
Figure 2: Cerebrospinal (CSF) -ACTH, -glutamate, -NGF and -IGF-1 in patients with Rett syndrome (RTT) and controls: ** CSF-ACTH 0.009, *** CSF-Glutamate 0.0006, ** CSF-NGF 0,001.
Table 3: Comparisons of concentrations of biochemical parameters in patients with RTT who were less than or older than 8 years of age. Data are mean (SD).
|
Parameter |
Age < 8 years n=9 |
Age > 8 years n=6 |
P-value |
|
CSF-ACTH ng/ml |
33.6 (7.6) |
29.8 (6.2) |
0.56 |
|
CSF-glutamate nmol/l |
384 (132) |
320 (72) |
0.54 |
|
CSF-IGF-1 µg/l |
0.39 (0.18) |
0.56 (0.19) |
0.24 |
|
CSF-ACTH/glutamate-ratio |
0.11 (0.05) |
0.09 (0.03) |
1.0 |
|
CSF-IGF-1/glutamate-ratio |
0.0012 (0.0005) |
0.0017 (0.0008) |
0.31 |
|
CSF-Acetylcholinesterase activity nmol/mg prot/minute |
81.1 (30.7) |
73.2 (30.6) |
0.54 |
|
CSF-Nerve growth factor pg/ml |
3.24 (3.29) |
0.74 (1.27) |
0.31 |
CSF=cerebrospinal fluid, ACTH=adrenocorticotropic hormone, IGF= insulin-like growth factor
V CSF-Acetylcholinesterase
CSF-acetylcholinesterase was calculated in proportion to the amount of CSFprotein. In controls, it varied between 7.0 and 20.1 nmol/ml/minute (mean 15.1, SD 5.0) and between 22.8 and 106.5 nmol/mg prot/minute (57.0, SD 23.8). Cholinesterase activity in patients with RTT varied between 9.9 and 29.8 nmol/ml/minute (mean 16.2, SD 5.9) and between 53.9 and 135.8 nmol/mg prot/minute (77.6, SD 29.4) (p=0.12). There were no significant differences patients with RTT who had epilepsy (n=8) compared those that did not (n=4; p=0.93) and those receiving carbamazepine (n=2) or valproate (n=4). There were no significant molecular changes in the other parameters in early /late onset groups (Table 2) or in the groups of patients with age less/more than 8 years (Table 3).
VI Outcomes
Five of the early onset patients could never walk. One (Patient 6) learnt to walk at the age of 20 months but soon lost this ability. All eight late onset patients learnt to walk at the age of 12 to 21 months. Moderate to severe hypo densities in SPECT were associated with early deceleration and poor outcome (no gait) in all cases. In our study, the outcome predictors were seen already at an early age (between one and four years in 8 cases). The patients with poor outcome (gross motor disability) differed from those with a favorable outcome by the early onset of the symptoms, early deceleration of head growth, and moderate to severe hypo perfusion in SPECT studies in the frontal lobe.
Discussion
Extensive studies were performed to search for pathogenic factors. The results of the present study are unique because CSF studies are not done for RTT. The aim of this study was to evaluate possible predictive factors from this cohort combining our earlier and new data. Predictive values of the early clinical signs of RTT were sought in the British Isles Survey for RTT, registering 1159 cases over 20 years. Early severity scores (based on muscle tone, locomotor ability, feeding difficulty, scoliosis and epilepsy) were highly significantly associated with the severity of later outcome and reduced cumulative survival [19]. We have these early severity scores in clinical data basis but selected the simplest measures: locomotor activity (gait) for evaluation because muscle tone is a difficult to measure in RTT patients, feeding difficulty (gastrostomy was not often used at that time) as well as scoliosis (young age of our patients) were also difficult measures. Epilepsy was seen in almost all our patients and seizures were difficult to quantify. In the British study, the mutations T158M, R255X and R168X were generally associated with more severe and R306C and R133C with less severe disease [19]. In our present study five of our nine patients with favorable outcome had mutations at 306C. In one recent large studies from USA, individuals with the R168X mutations were more severely affected than those with R294X, and in another p.Arg106Trp, p.Arg168X, p.Arg255X and p.Arg270X were more affected than p.Arg133Cys, p.Arg294X and p.Arg306Cys truncations [20, 21]. However, exceptions made these unreliable predictors of outcome.
I Head Growth and Motor Disability
A major and consistent abnormality in RTT is reduced brain size and weight [22]. Our main finding was that the most reduced brain size with early deceleration of head growth occurred in patients with early onset of symptoms and with unfavorable outcome. A review of brain weights at autopsy in patients of different ages revealed no evidence of progressive disease in brain weight at different ages [23]. This was supported by MRI studies [24]. In a longitudinal study, there was a strong relationship between gross motor function and the degree of head growth deceleration at the ages of 6 and 12 years [25]. Our study showed that this prognostic sign can be seen already in an early age.
II Neuronal Growth Factors and SPECT
Nerve growth factor is important for cholinergic neurons of the frontal brain. Nerve growth factor activates the gene expression of acetylcholinesterase, the enzyme responsible for acetylcholine formation [26]. It prevents degeneration of basal forebrain cholinergic neurons. In RTT, the cholinergic system is more severely affected than other cortical neurons [26, 27]. Low levels of NGF in CSF and postmortem brain tissue are reported [28]. Consistent to that, in our earlier studies of RTT, we have shown that CSF-NGF was low [5]. The early onset group also showed moderate or severe hypoperfusion defects, particularly in the frontal lobe compared to the late onset-group. In the late onset-group alteration in brain perfusion was no such evident
III CSF ACTH/Glutamate
The involvement of excitatory amino acids in the control of ACTH release is well established in clinical studies. ACTH-related peptides have neurotrophic and neuroprotective actions, and glutamate excitatory properties [29]. Pharmacological interventions influencing interaction between glutamate and the hypothalamic-pituitary-adrenal axis are promising treatment possibilities in psychiatry and neurology [30]. CSF levels of glutamate (p=0.0006) and ACTH (p= 0.009) were higher in patients with RTT compared to controls. The activation of ionotropic glutamate receptors has a stimulatory effect on ACTH release [30]. Thus, one explanation for the high CSF-ACTH in our patients could be the stimulatory effect of glutamate. CSF-ACTH/glutamate-ratio was significantly higher in the early onset-group compared to the late onset-group (p=0.032). This may represent an attempt by damaged brain to restore the balance between excitatory and inhibitory transmission because of neurochemical plasticity.
IV CSF-Glutamate and Biphasic Clinical Condition?
RTT is among the chronic neurologic diseases in which neuronal damage is thought to be mediated, at least in part, by stimulation of glutamate receptors [31, 32]. The developing brain is particularly vulnerable to NMDA receptor overstimulation [33]. Both too little and too much activity at NMDA receptors can damage the developing brain [1]. CSF concentrations of glutamate were significantly higher in our patients with RTT compared to controls [3]. Epilepsy occurred in most of our patients. However, patients without epilepsy also exhibited high levels of glutamate in the CSF, which supports the conclusion that epilepsy alone cannot account for high levels of CSF-glutamate in RTT patients. Because both patients in early and late onset group had epilepsy, we could not show whether CSF-glutamate differences would reflect biphasic clinical conditions; clinical signs of excitatory activity (seizures early in childhood ) but reduced signs of excessive CNS activity later in childhood as suggested by other studies [9, 10, 12].
V Cholinergic Transmitters
As a marker of cholinergic dysfunction, we examined the activity of cholinesterase. As far as we know, no similar studies have been done earlier. Furthermore, we wanted to see whether the type of MECP2 mutation would be in related to cholinesterase activity. We expected to find a difference in acetylcholinesterase between the patients with favorable and unfavorable outcomes reflecting magnitude of the disturbance in the brain. The two patients in whom the activities were highest were the most mildly affected, especially regarding motor function. The most affected children (early onset-group) had moderate to severe hypo perfusion in frontal brain. We could not find any significant differences in early or late onset groups which might be due to small number of patients.
VI Glutamate/IGF-1 Ratio
We determined the ratio of CSF levels of glutamate to IGF-1 as a possible marker for favorable/unfavorable outcomes. Growth factors alleviate NMDA toxicity, which is needed to bring balance between excitation and inhibition [34-36]. IGF-1 has been reported to be deficient in MECP2-knock-out mice [37]. Several clinical studies have shown possible efficacy of IGF-1 in attempting to normalize synaptic abnormalities in disorders of the CNS [38, 39]. Preliminary study in girls with RTT showed that IGF-1 is well tolerated and shows some benefit [38]. Very recently, double-blind, randomized, placebo-controlled study of trofinitide (analog of the amino-terminal tripeptide of IGF-1 in pediatric RTT provided evidence that trofinitide is a potentially viable treatment for the core signs and symptoms of RTT [40]. Eighty-two children aged 5 to 15 years were randomized..
Limitations of the Study
Because all the studies could not be done for all patients (small samples, refusal of the parents, severe scoliosis, etc.) the series presented here has certain limitations. There was no data available regarding why some patients with RTT had normal development for longer and progression of the disease is over a longer period, or what triggered the progress of the disease. There might have been genetic differences. Some cells which maintain synapses may be more vulnerable than the others to disturbances in metabolism. In our small study, we could not find any explanation for these differences. Moreover, our control children did not all have normal development. However, in another our study, we had 16 healthy children (mean age 7 years (range 1 mo-15 years) admitted for surgery on the lower part of the body to be performed under spinal anesthesia in a control-group for children with autism [41]. These children had similar CSF-IGF-1 and CSF- and serum-BDNF concentrations to the control children in this study.
Conclusion
The patients with RTT who had poor outcome differed from those with more favorable outcomes showed early onset of the symptoms, early deceleration of head size, and moderate to severe hypoperfusion in the frontal lobes in SPECT, corresponding low CSF-NGF found in patients with RTT; NGF being important for the cholinergic activity of the forebrain. We could not find any significant molecular biomarkers for predictive outcome in our small number of patients. However, CSF-ACTH/glutamate-ratio was higher in early onset RTT with unfavorable outcome than in late onset-patients with favorable outcome. This might mean an attempt to restore the balance between excitatory and inhibitory transmission. Certain genes were associated with favorable outcome. Clinical parameters were in our study the most predictive poor outcome measures.
Funding
None.
Conflicts of Interest
None.
Author Contribution
All authors have made substantial contributions to all of the following: (i) the conception or design of the study or the acquisition, or analysis and interpretation of data of the work, (ii) drafting the article or revising it critically for intellectual content, and (iii) final approval of the version to be submitted.
Accessibility Statement
The article`s supporting main data and materials can be accessed in the Helsinki University. The investigations were made at the Hospital for Children and Adolescents, Helsinki University, Helsinki, Finland where the original data is kept.
Ethics
The study adhered to the tenets of the Helsinki Declaration. The Ethics Committee of the Children`s Hospital, Helsinki approved the study. The study had institutional approval.
Consent
Patients or their caregivers gave informed consent to the research and to publications of the results.
Article Info
Article Type
Research ArticlePublication history
Received: Mon 30, Dec 2019Accepted: Thu 16, Jan 2020
Published: Fri 24, Jan 2020
Copyright
© 2023 Raili Riikonen. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited. Hosting by Science Repository.DOI: 10.31487/j.NNB.2020.01.01
Figures & Tables
Table 1: Patients’ characteristics. Comparison of early onset-group to late onset-group, p-values Mann-Whitney-test.
|
|
Normal mo |
HC (SD) |
Epilepsy yrs:mo |
AED |
CSF ACTH |
CSF Glutam |
CSF IGF-1 |
CSF Acetyl |
CSF NGF |
Gene |
Gait mo |
SPECT |
|||
|
yrs:mo |
Front |
Temp |
Par |
||||||||||||
|
Early onset 6-8 months, N=6 |
|||||||||||||||
|
#1 / 1.7 yrs |
6 |
-2 |
No |
No |
43 |
265 |
0.53 |
97.7 |
0.1 |
R255X |
No |
|
R++/ L+++ |
R+ |
|
|
#2 / 4.2 yrs. |
6 |
-3.5 |
No |
No |
ND |
407 |
0.56 |
ND |
6.6 |
R270X |
No |
4:2 |
R+++/ L++ |
T++ |
R++/ L+ |
|
#3 / 4.2yrs |
6 |
-2.5 |
1:8 |
VPA |
25 |
213 |
0.16 |
72.5 |
0.16 |
G269X |
No |
ND |
|
|
|
|
#4 / 4.2 yrs |
6 |
ND |
1:6 |
VPA |
|
|
|
ND |
|
|
No |
4:2 |
R+++/L++ |
TR+++ |
|
|
#5 / 17.4 yrs |
8 |
-3.0 |
1:7 |
VPA |
36 |
252 |
0.44 |
56.0 |
0.1 |
ND |
No |
17:5 |
R+/L++ |
|
L+ |
|
#6 /11.4 yrs |
5 |
-3.0 |
4:4 |
VPA |
34 |
436 |
0.61 |
55.1 |
0.1 |
R302A |
20 / later lost |
11:6 |
Hypometabolism |
||
|
Late onset 10-24 months, N=9 |
|||||||||||||||
|
#7 / 16.5 yrs |
12 |
-0.8 |
2:6 |
CBZ/VPA |
26 |
332 |
0.29 |
ND |
0.4 |
R306C |
18 |
ND |
|
|
|
|
#8 / 11.3yrs |
10 |
-1.5 |
4:0 |
CBZ |
ND |
ND |
0.74 |
66.2 |
0.1 |
R306C |
21 |
11:4 |
Normal |
||
|
#9 / 2.9 yrs |
15 |
-2 |
No |
No |
31 |
521 |
0.47 |
60.3 |
0.1 |
R106W |
14 |
3:2 |
|
R+ |
|
|
#10 / 3yrs.11mo |
15 |
-0.3 |
1:6 |
VPA |
40 |
539 |
0.48 |
53.9 |
ND |
R306C |
20 |
3:5 |
R++/L++ |
|
L+ |
|
#11 /6.5 yrs. |
23 |
-1.5 |
4:0 |
VPA |
ND |
361 |
ND |
ND |
6.7 |
R306C |
20 |
ND |
|
|
|
|
#12 / 9.8 yrs |
18 |
-1.7 |
3:7 |
CBZ |
23 |
311 |
0.48 |
61.4 |
3.0 |
R306C |
16 |
9:10 |
R+ |
|
R+ |
|
#13 / 2.3 yrs |
72 |
-1.3 |
No |
No |
ND |
ND |
0.16 |
135.8 |
2.7 |
R294X |
12 |
2:7 |
R+ |
|
|
|
#14 / 13.4 yrs |
18 |
-0.8 |
2:0 |
CBZ / VPA |
ND |
270 |
0.79 |
127.4 |
0.1 |
R294X |
16 |
13:5 |
|
R+/L+ |
|
|
#15 / 2 yrs |
ND |
0 |
No |
No |
29 |
|
|
66.3 |
|
T158M |
20 |
ND |
|
|
|
|
P-value |
|||||||||||||||
|
early vs. late |
|
0.002 |
|
|
0.41 |
0.25 |
1.0 |
0.51 |
0.53 |
|
|
|
|
|
|
Abbreviations: mo=months, yrs=years, HC: head circumference, AED: antiepileptic drugs, ACTH: adrenocorticotrophic hormone, CSF: cerebrospinal fluid, IGF-1: insulin-like growth factor-1, Glutam: glutamate, Acetyl: acetylcholine esterase; NGF: beta nerve growth factor, II-IV: Stages of Rett syndrome; ND: :no data, Front: frontal lobe; Temp: temporal lobe; Par: parietal lobe; R:right; L:left, + = slight; ++ = moderate, +++ = severe; N: normal.
Table 2: Comparisons of concentrations of biochemical parameters in patients with an early (regression of development before age of 5-8 months) versus late onset (regression of development at age of 10-24 months) of RTT. Data are mean (SD).
|
Parameter |
Early onset n=6 |
Late onset n=9 |
P-value |
|
CSF-ACTH ng/ml |
34.5 (7.4) |
29.8 (6.5) |
0.41 |
|
CSF-glutamate nmol/l |
315 (100) |
389 (113) |
0.25 |
|
CSF-IGF-1 µg/l |
0.46 (0.18) |
0.49 (0.22) |
1.0 |
|
CSF-ACTH/glutamate-ratio |
0.11 (0.03) |
0.07 (0.01) |
0.032 |
|
CSF-IGF-1/glutamate-ratio |
0.0014 (0.0005) |
0.0014 (0.0009) |
0.84 |
|
CSF-Acetylcholinesterase activity nmol/mg prot/minute |
70.3 (19.9) |
81.6 (34.5) |
0.79 |
|
CSF-Nerve growth factor pg/ml |
2.27 (3.75) |
1.87 (2.48) |
0.53 |
CSF=cerebrospinal fluid, ACTH=adrenocorticotropic hormone, IGF= insulin-like growth factor.
Table 3: Comparisons of concentrations of biochemical parameters in patients with RTT who were less than or older than 8 years of age. Data are mean (SD).
|
Parameter |
Age < 8 years n=9 |
Age > 8 years n=6 |
P-value |
|
CSF-ACTH ng/ml |
33.6 (7.6) |
29.8 (6.2) |
0.56 |
|
CSF-glutamate nmol/l |
384 (132) |
320 (72) |
0.54 |
|
CSF-IGF-1 µg/l |
0.39 (0.18) |
0.56 (0.19) |
0.24 |
|
CSF-ACTH/glutamate-ratio |
0.11 (0.05) |
0.09 (0.03) |
1.0 |
|
CSF-IGF-1/glutamate-ratio |
0.0012 (0.0005) |
0.0017 (0.0008) |
0.31 |
|
CSF-Acetylcholinesterase activity nmol/mg prot/minute |
81.1 (30.7) |
73.2 (30.6) |
0.54 |
|
CSF-Nerve growth factor pg/ml |
3.24 (3.29) |
0.74 (1.27) |
0.31 |
CSF=cerebrospinal fluid, ACTH=adrenocorticotropic hormone, IGF= insulin-like growth factor
References
- Johnston M, Blue ME, Naidu S (2015) Recent advances in understanding synaptic abnormalities in Rett syndrome. F1000Res 4: pii: F1000 Faculty Rev-1490. [Crossref]
- Vanhala, R (1998) Rett syndrome. A search for etiopathogenetic factors. Academic Dissertation, University of Helsinki, Helsinki, Finland.
- Lappalainen R, Riikonen R (1996) High levels of cerebrospinal fluid glutamate in Rett syndrome. Pediatr Neurol 15: 213-216. [Crossref]
- Vanhala R, Turpeinen U, Riikonen R (2000) Insulin-like growth factor-1 in cerebrospinal fluid and serum in Rett syndrome. J Child Neurol 15: 797-802. [Crossref]
- Lappalainen R, Lindholm D, Riikonen R (1996) Low levels of nerve growth factor in cerebrospinal fluid of children with Rett syndrome. J Child Neurol 11: 296-300. [Crossref]
- Vanhala R, Korhonen L, Mikelsaar M, Lindholm D, Riikonen R (1998) Neurotrophic factors in cerebrospinal fluid and serum in patients with Rett syndrome. J Child Neurol 13: 429-433. [Crossref]
- Khwaja OS, Ho E, Barnes KV, O'Leary HM, Pereira LM et al. (2014) Safety, pharmacokinetics, and preliminary assessment of efficacy of mecasermin (recombinant human IGF-1) for treatment of Rett syndrome. Proc Natl Acad Sci USA 111: 4596-4601. [Crossref]
- Hagberg B (1993) Clinical criteria, stages and natural history. In: Rett syndrome–Clinical and biological aspects (ed.) Hagberg B, Mac Keith Press, London 4-20.
- Blue ME, Naidu S, Johnston MV (1999) Development of amino acid receptors in frontal cortex from girls with Rett syndrome. Ann Neurol 45: 541-545. [Crossref]
- Blue ME, Naidu S, Johnston MV (1999) Altered development of glutamate and GABA receptors in basal ganglia of girls with Rett syndrome. Exp Neurol 156: 345-52. [Crossref]
- Horská A, Farage L, Bibat G, Nagae LM, Kaufmann WE et al. (2009) Brain metabolism in Rett syndrome: Age, clinical, and genotype correlations. Ann Neurol 65: 90-97. [Crossref]
- Blue ME, Kaufmann WE, Bressler J, Eyring C, O'driscoll C et al. (2011) Temporal and regional alterations in NMDA receptor expression in Mecp2-null mice. Anat Rec (Hoboken) 294: 1624-1634. [Crossref]
- Lappalainen R, Liewendahl K, Sainio P, Riikonen RS (1997) Brain perfusion SPECT and EEG findings in Rett syndrome. Acta Neurol Scand 95: 44-50. [Crossref]
- Soininen HS, Jolkkonen JT, Reinikainen KJ, Halonen TO, Riekkinen PJ (1984) Reduced cholinesterase and somatostatin-like immunoreactivity in the cerebrospinal fluid of patients with dementia of Alzheimer type. J Neurol Sci 63: 167-172. [Crossref]
- Valtonen P, Haapalinna A, Riekkinen P, Halonen T (1995) Effect of alfa-2-adrenergic dexmedetomidine and atipamezole on extracellular amino acid levels in vivo. Eur J Pharmacol 285: 239-246. [Crossref]
- Lindholm D, Castren E, da Penha Berzagi M, Thoenen H (1993) Effects of neurotransmitters and hormones on neuronal production of neurotrophins. Seminar Neurosci 5: 279-283.
- Auranen M, Vanhala R, Vosman M, Levander M, Varilo T et al. (2001) MECP2 gene analysis in classical Rett syndrome and in patients with Rett-like features. Neurology 56: 611-617. [Crossref]
- Amir RE, Van den Veyver IB, Schultz R, Malicki DM, Tran CQ et al. (2000) Influence of mutation type and X chromosome inactivation on Rett syndrome phenotypes. Ann Neurol 9: 670-679. [Crossref]
- Kerr A, Prescott R (2005) Predictive value of the early clinical signs in Rett disorder. Brain Dev 27: S20-S24. [Crossref]
- Neul JL, Fang P, Barrish J, Lane J, Caeg E et al. (2008) Specific mutations in Methyl-CpG-binding protein 2 confer different severity in Rett syndrome. Neurology 15: 1313-1321. [Crossref]
- Cuddapah VA, Pillai RB, Shekar KV, Lane JB, Motil KJ et al. (2014) Methyl-CpG-binding protein 2 (MCP2) mutation type is associated with disease severity in Rett syndrome. J Med Genet 51: 152-158. [Crossref]
- Naidu S (1997) Rett syndrome: A disorder affecting early brain growth. Ann Neurol 42: 3-10. [Crossref]
- Armstrong DD (1995) The neuropathology of Rett syndrome--overview 1994. Neuropediatrics 26: 100-104. [Crossref]
- Subramaniam B, Naidu S, Reiss A (1997) Neuroanatomy in Rett syndrome: cerebral and posterior fossa. Neurology 48: 399-407. [Crossref]
- Stenbom Y, Engerström IW, Hagberg G (1995) Gross motor disability and head growth in Rett syndrome. Neuropediatrics 26: 85-86. [Crossref]
- Hohmann CF, Brooks AR, Coyle JT (1988) Neonatal lesions of the basal forebrain cholinergic neurons result in abnormal cortical development. Brain Res 43: 253-264. [Crossref]
- Wenk GL, Hauss Wegrzyniak B (1999) Altered cholinergic function in the basal forebrain of girls with Rett syndrome. Neuropediatrics 30: 125-129. [Crossref]
- Lipani JD, Bhattacharjee MB, Corey DM, Lee DA (2000) Reduced nerve growth factor in Rett syndrome in postmortem brain tissue. J Neuropathol Exp Neurol 59: 889-895. [Crossref]
- Hol EM, Gispen WH, Bär PR (1995) ACTH-related peptides: Receptors and signal transduction systems involved in their neurotrophic and neuroprotective actions. Peptides 16: 979-993. [Crossref]
- Jezova D (2005) Control of ACTH secretion by excitatory amino acids functional significance and clinical implications. Endocrine 28: 287-293. [Crossref]
- Lipton S, Rosenberg PA (1994) Excitatory amino acids as a final common pathway for neurologic disorders. N Engl J Med 330: 613-622. [Crossref]
- Maezawa I, Jin LW (2010) Rett syndrome microglia damage dendrites and synapses by elevated release of glutamate. J Neurosci 30: 5346-5356. [Crossref]
- Johnston MV, Hohmann C, Blue ME (1995) Neurobiology of Rett syndrome. Neuropediatrics 26: 119-122. [Crossref]
- Escartin C, Boyer F, Benelmans AP, Hantraye P, Brouilet E (2004) Insulin growth factor-1 protects against neurotoxcity in rat striatum. Neuroreport 15: 2251-2254. [Crossref]
- Wood TL, Loladze V, Altieri S, Gangoli N, Levison SW et al. (2007) Delayed IGF-1 administration rescues oligodendrocyte progenitors from glutamate-induced cell death and hypoxic-ischemic brain damage. Dev Neurosci 29: 302-310. [Crossref]
- Wang Y, Wang W, Li D, Li M, Wang P et al. (2014) IGF-1 alleviates NMDA-induced excitotoxicity in cultured hippocampal neurons against autophagy via the NR2B/PI3K-AKT-mTOR pathway. J Cell Physiol 229: 1618-1629. [Crossref]
- Castro J, Garcia RI, Kwok S, Banerjee A, Petravicz J et al. (2014) Functional recovery with recombinant human IGF1 treatment in a mouse model of Rett syndrome. Proc Natl Acad Sci 111: 99441-99446. [Crossref]
- Costales J, Kolevzon A (2016) The therapeutic potential of insulin-like growth factor-1 in central nervous system disorders. Neurosci Biobehav Rev 63: 207-222. [Crossref]
- Riikonen R (2016) Treatment of autistic spectrum disorder with insulin-like growth factors. Eur J Paediatr Neurol 20: 816-823. [Crossref]
- Glaze DG, Neul JL, Kaufmann WE, Berry Kravis E, Condon S et al. (2019) Double-blind, randomized, placebo-controlled study of trofenitide in pediatric Rett syndrome. Neurology 92: e1912-e1925. [Crossref]
- Riikonen R, Makkonen I, Vanhala R, Turpeinen U, Kuikka J et al. (2006) Cerebrospinal fluid insulin-like growth factors IGF-1 and IGF-2 in infantile autism. Dev Med Child Neurol 48: 753-755. [Crossref]