Potential Mechanisms Involved in Diabetic Teratogenesis During Embryonic Neural Tube Closure
A B S T R A C T
Diabetes mellitus during pregnancy is a risk factor for improper closure of the neural tube during embryonic development. It is likely that this occurs via several parallel pathways. Previously proposed pathways for diabetic teratogenesis during neural tube closure include augmented production of nitric acid; reduction of Pax3; and increased production of long-chain acyl-CoA synthetase 1 (ACSL1). In the current discussion, an additional hypothesis is offered: that hyperglycemia might exert its teratogenic effects on the developing embryo via overexpression of collagen IV, leading to collagen imbalance and errors of remodelling.
Keywords
Neural tube defects, diabetes mellitus, macrophages, collagen
Introduction
Diabetes mellitus during pregnancy is a known risk factor for imperfect closure of the developing embryonic neural tube. The diabetic state most likely increases risk for neural tube defects via multiple parallel pathways. Previously proposed pathways for diabetic teratogenesis during neural tube closure have included augmented production of nitric acid, which is toxic in excess to the developing neural tube; reduction of Pax3, which suppresses p53 dependent cell death during embryonic development of the neural tube; and increased production of long-chain acyl-CoA synthetase 1 (ACSL1), which promotes polarization of macrophages or toward a more inflammatory phenotype. These mechanisms have been summarized in a previous analysis [1].
It is of interest that all of the three mechanisms suspected to be involved in diabetic teratogenesis above -- Pax3 reduction, nitric acid excess, and ACSL1 upregulation -- should reduce the population of macrophages belonging the M2 or M2-like phenotype (Hereafter, “M2” includes “M2-like” in the text below.) A reduction in Pax3 would be expected to result in fewer M2 macrophages via promotion of p53 associated polarization to the M1 phenotype. Increased production of nitric acid should result in fewer M2 macrophages via peroxynitrite associated macrophage apoptosis. Upregulation of ACSL1 would be expected to result in fewer macrophages via the pro-inflammatory effect of ACSL1 on macrophages. M2 macrophages, in turn, are associated with regulation and deposition of structural proteins such as collagen and actin, molecules which play significant roles in closure of the neural tube [1]. Circumstantial evidence suggests that these mechanisms may influence the structure of the developing neural tube by virtue of, or in connection to, their effects on the size of the M2 macrophage pool.
M2 macrophages as a central player in neural tube closure
In a previous analysis, our hypothesis was that M2 macrophages might play a role of “middle man” between known protective factors (folic acid, other factors involved in one carbon metabolism, inositol, and signalling molecules such as inositol trisphosphate and sphingosine-1-phosphate) and the structural work of the developing embryo. As was discussed in that analysis, in order to demonstrate that M2-like macrophages play a role in influencing neural tube structure, one would need to demonstrate that M2-like macrophages can exert effects, either directly or indirectly, on factors such as actin, collagen, integrin, and others -- factors which contribute to the structural integrity of the embryo. A “proof” for such a relationship has not yet been established: testing is needed. Nevertheless, since M2 macrophages have already been demonstrated to play a role in the remodelling of collagen in other tissues, it is tempting to implicate M2 macrophages in the regulation and remodelling of collagen in the developing embryo, as well [1].
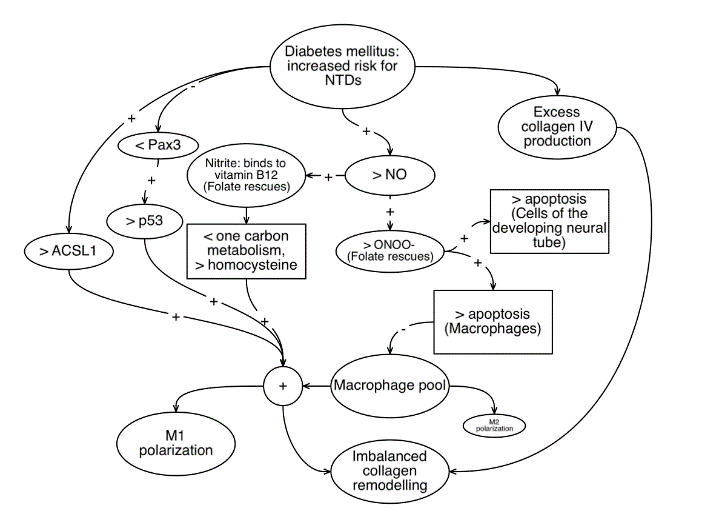
Figure 1: Illustrates various potential parallel mechanisms by which diabetes mellitus might interfere with collagen remodelling and neural tube closure in the developing embryo. It is hoped that the various hypotheses and associations noted above will be further clarified through experimentation on embryonic tissue.
A potential role for M2 macrophages in collagen IV remodelling
One type of collagen, collagen IV, has been implicated in the development of the neural plate in the mouse embryo [2]. One would expect that, under normal conditions, collagen IV deposition and removal would be an ongoing, continuous process. Such remodelling would be expected to occur throughout embryonic development. During folding and closure of the neural tube, attainment of optimal geometry might involve even more resorption and breakdown of collagen. Thus, it is perhaps not so surprising that a deficit of folic acid, the best-known preventative factor for neural tube defects, results in an increased accumulation of collagen IV in some tissues [3].
In addition to folic acid, macrophages have been implicated in collagen and folic acid related pathways. Folic acid activates the macrophage folic acid receptor beta (FR-beta), a receptor which is heavily expressed on activated macrophages polarized to the M2 phenotype. The M2 macrophage FR-beta receptor, in turn, regulates macrophage attachment to collagen. Thus, folic acid deficiency has the potential to impair adhesion of macrophages to collagen, and M2 macrophages have been found to mediate collagen remodelling and degradation [4-6]. It is possible that M2 macrophages are players which translate the “input” of folic acid into the “output” of collagen IV remodelling during neural tube closure.
Disruption of collagen IV remodelling in diabetes mellitus
Similar to the effects of folic acid deficiency, in the present analysis, it is suggested that diabetic teratogenesis might also involve disruption of collagen IV remodelling during neural tube closure. The evidence is limited but suggestive. It has been known for some time that the diabetic state predisposes to increased urinary excretion of collagen IV, and that urinary excretion of collagen IV is even higher in those with diabetic nephropathy [7, 8]. In vitro, a hyperglycemic environment upregulates expression of collagen IV synthesis, potentially leading to collagen IV accumulation [9]. Thus, it is possible that diabetes mellitus predisposes to collagen IV imbalance with resultant impaired collagen IV remodelling
Conflicts of Interest
The author acknowledges no conflicts of interest.
Funding
The author acknowledges no sponsors or sources of funding.
Article Info
Article Type
Research ArticlePublication history
Received: Wed 04, Sep 2019Accepted: Mon 23, Sep 2019
Published: Mon 30, Sep 2019
Copyright
© 2023 Janice Block. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited. Hosting by Science Repository.DOI: 10.31487/j.JDMC.2019.01.06
Figures & Tables
References
- Block J (2019) M2-like cells from the macrophage lineage might play a central role in closure of the embryonic neural tube. Med Hypotheses 129: 109264. [Crossref]
- O’Shea KS (1987) Differential deposition of basement membrane components during formation of the caudal neural tube in the mouse embryo. Development 99: 509-519. [Crossref]
- Sijilmassi O, Lopez Alonso JM, Barrio Asensio MC, Del Rio Sevilla A (2018) Collagen IV and laminin-1 expression in embryonic mouse lens using principal components analysis technique. J Microsc 271: 207. [Crossref]
- Machacek C, Supper V, Leksa V, Mitulovic G, Spittler A et. al. (2016) Folate Receptor β Regulates Integrin CD11b/CD18 Adhesion of a Macrophage Subset to Collagen. J Immunol 197: 2229-2238. [Crossref]
- Hautvast JG, Barnes MJ (1974) Collagen metabolism in folic acid deficiency. Br J Nutr 32: 457-469. [Crossref]
- Madsen DH, Leonard D, Masedunskas A, Moyer A, Jürgensen HJ et. al. (2013) M2-like macrophages are responsible for collagen degradation through a mannose receptor-mediated pathway. J Cell Biol 202: 951-966. [Crossref]
- Morita M, Hanai K, Uchigata Y (2014) Urinary type IV collagen as a predictor for the incidence of microalbuminuria in young patients with Type 1 diabetes. Diabet Med 31: 213. [Crossref]
- Sthaneshwar P, Chan SP (2010) Urinary type IV collagen levels in diabetes mellitus. Malays J Pathol 32: 43. [Crossref]
- Tahara A, Tsukada J, Tomura Y, Yatsu T, Shibasaki M (2012) Effects of high glucose on AVP-induced hyperplasia, hypertrophy, and type IV collagen synthesis in cultured rat mesangial cells. Endocr. Res 37: 216-227. [Crossref]