Pediatric Sinonasal Rhabdomyosarcoma: Clinical Characteristics and Surgical Role
A B S T R A C T
Introduction: Rhabdomyosarcoma is the most common pediatric soft tissue sarcoma. It represents 5-8% of pediatric tumors. Head and neck is considered the most common site of RMS origin (40%). Parameningeal, orbital and non parameningeal RMS approximately represent 50%, 25% and 25% respectively. Histopathological evaluation demonstrates small round cells with high cytological variability which stain positive with desmin, myogenin and MyoD1. The mainstay treatment of RMS depends on chemotherapy and radiation therapy with a limited role of surgery.
Method: A retrospective chart review for pediatric sinonasal rhabdomyosarcoma diagnosed and managed at King Fahad Specialist Hospital, Dammam, Saudi Arabia, and a literature review of pediatric sinonasal rhabdomyosarcoma was conducted.
Results: A total of four cases were identified for the period (2011-2017), and a thorough review of their medical records and radiological imaging were done.
Conclusion: Pediatric sinonasal rhabdomyosarcoma may initially present with symptoms mimicking rhinosinusitis. Biopsy and histological evaluation are the most essential steps to exclude malignancy. Intergroup Rhabdomyosarcoma Studies (IRS) established a staging system based on tumor extension and resectability. The role of surgery in RMS may be limited for obtaining biopsies for diagnosis and for palliative purposes. Popular antineoplastic agents used to treat RMS include vincristine, cyclophosphamide, actinomycin D, and adriamycin. Chemotherapy with alkylating agents has achieved a relapsed free survival 90% for nonparameningeal tumors and 65% for parameningeal tumors.
Keywords
Rhabdomyosarcoma, pediatrics, sinonasal, paransasl sinuses
Introduction
Rhabdomyosarcoma is considered to be the most common soft tissue sarcoma among the pediatric population [1, 2]. It is a rare tumor, representing 5-8% of pediatric tumors [3]. RMS behaves as an aggressive malignant tumor and has a tendency of early metastasis [4]. Variable subtypes include embryonal, alveolar, botryoid variant, spindle cell embryonal and anaplastic, showed prognostic values [5]. Nevoid basal cell carcinoma (Gorlin syndrome), Neurofibromatosis type 1, Beckwith- Wiedemann syndrome and Rubinstein-Taybi syndrome, p53 tumor suppressor gene mutations and rhabdomyosarcoma oncogene mutations have been associated with rhabdomyosarcoma [6-9]. Head and neck is considered the most common site of RMS origin (40%). Parameningeal, orbital and non parameningeal RMS approximately represent 50%, 25% and 25% respectively [10, 11]. The initial symptoms of pediatric sinonasal rhabdomyosarcoma may mimic rhinosinusitis and delay diagnosis. Comprehensive clinical evaluation with the detailed history and physical examination is crucial. The endoscopic and radiological evaluation must be performed before obtaining a biopsy of a unilateral nasal mass. Embryonal and alveolar histological subtypes represent the majority of pediatric rhabdomyosarcoma [12]. The histopathological evaluation usually demonstrates small round cells with high cytological variability. Immunohistochemical staining helps to confirm the diagnosis. RMS stained positive with desmin, myogenin and MyoD1. Cytokeratin, S-100 and epithelial membrane antigen do not stain, are negative, and help to exclude the other differential diagnoses for RMS [13]. The mainstay treatment of RMS depends on chemotherapy and radiation therapy with the limited role of surgery [14]. In this article, we present a case series of pediatric rhabdomyosarcoma involving paranasal sinuses, in addition to the clinical approach and treatment.
Method
A retrospective chart review for pediatric sinonasal rhabdomyosarcoma diagnosed and managed at King Fahad Specialist Hospital, Dammam, Saudi Arabia, was conducted after IRB committee approval. A total of 4 cases were identified for the period (2010 -2017), and a thorough review of their medical records and radiological imaging were done. A literature review of pediatric sinonasal rhabdomyosarcoma was conducted.

Figure 1: Endoscopic view of right nasal cavity shows a huge fleshy mass, obstructing choana and nasopharynx.
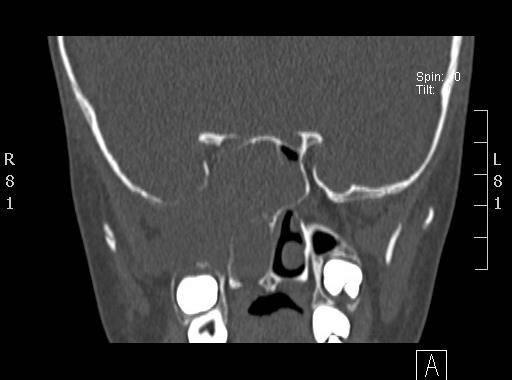
Figure 2: CT scan of paranasal sinuses, showing right sided lesion involving posterior ethmoids and sphenoid with extension to infratemporal fossa and dehiscent skull base at roof of sphenoid.
Results
Case 1
An eight-year-old boy presented to the rhinology clinic with a complaint of nasal obstruction, rhinorrhea, and headache. The patient’s symptoms progressed over a month before referral to our hospital. He developed right eye ptosis and double vision. Nasal endoscopy showed a fleshy mass in the right nasal cavity (Figure 1) involving right sphenoid sinus, posterior ethmoids obstructing right choana and nasopharynx. Computed tomography (CT) of the nose and paranasal sinuses demonstrated a right sided sphenoid and posterior ethmoids lesion (Figure 2), extending to the infratemporal area and nasopharynx with the area of dehiscent skull base at the sphenoid sinus roof.

Figure 3: MRI of paranasal sinuses showed a large soft tissue mass extend from greater wing of sphenoid to pterygoid plates.

Figure 4: MRI T1 with contrast showing enhanced lesion partially encases the cavernous portion of the right internal carotid artery.

Figure 5: PET/CT demonstrate intracranial extension to the right middle cranial fossa mainly through foramen ovale.
Magnetic Resonance images (MRI) of paranasal sinuses showed a large soft tissue mass involving the right side of the body of the greater wing of the sphenoid bone extending down to the right pterygoid plates and the right parapharyngeal area (Figure 3). The tumor was displacing the right lateral and medial pterygoid muscles, invading right sphenoid sinus and right posterior ethmoid air cells with scalloping of the posterior wall of maxillary antrum and extending to the nasopharynx. The tumor partially encases the cavernous portion of the right internal carotid artery (Figure 4). It appears iso-intense on T1WI, slightly high on T2WI with evidence of multiple signal voids centrally, which represent vessels. It was homogeneously enhancing post IV contrast administration and showed restriction on diffusion weighted images. The tumor showed intracranial extension to the right middle cranial fossa mainly through foramen ovale, as shown on PET CT scan (Figure 5). The patient underwent a biopsy of sinonasal mass under general anesthesia.

Figure 6: Microscopic view demonstrates sheets of small round blue cell (rhabdomyoblast) with slightly ample pink cytoplasm.

Figure 7: Microscopic view with positive myogenin and desmin stains, confirms diagnosis of embryonal rhabdomyosarcoma.
The intaraoperative frozen section showed small abnormal cells consistent with query malignancy. Final histopathological analysis reported infiltration of soft tissue and bone by small blue cells with abnormal chromatin and slightly ample pink cytoplasm (rhabdomyoblast) (Figure 6). Immunohistochemical staining showed strong staining with both myogenin and desmin (Figure 7) and negative staining with LCA, CD99 and Myo-D. Previous data were consistent with the diagnosis of right parameningeal embryonal rhabdomyosarcoma. The patient referred to the Pediatric Oncology service. Radiation therapy and chemotherapy consisting of VCR therapy (Vincristine, Dactinomycin and Cyclophosphamide), was initiated and tolerated by the patient. The patient responded well to therapy, and a follow-up imaging with MRI sinuses showed interval improvement of sinus disease and no sign of recurrence of the tumor.
Case 2
A nine-year-old girl presented with left eye proptosis and decreased vision few weeks prior to referral. The parents noticed their child complain from headache and a decreased oral intake. There was no clear history about nasal obstruction, rhinorrhea or epistaxis. Clinical examination revealed a left nasal and nasopharyngeal purple, fleshy mass. Left eye proptosis and decrease vision were evaluated by an ophthalmologist, defected left visual acuity was documented. A radiological evaluation with computed tomography and MRI was made. An extensive destructive lesion involved left posterior ethmoid sinus with eroded anterior skull base, intracranial extension and minimal extension to the left orbit at the superior medial aspect. The area of possible affection of the optic nerve was likely at the entrance of the optic canal bilaterally, where the lesion was minimally contacting the optic nerve. No obvious abnormal signal intensity within the optic nerve was seen or abnormal post IV contrast enhancement. The biopsy of left nasal cavity mass was done.
Histopathological evaluation showed malignant round blue cells, which demonstrated strong positive staining with desmin and myo-D. Further immunohistochemical staining with Pan-CK, CD45 and CD99 were negative. Morphological and immunohistochemical studies supported the diagnosis of rhabdomyosarcoma. Pediatric oncology referral and staging workup were conducted. RMS stage 4 was confirmed, and the patient started radiation and chemotherapy protocol. The patient is disease free on remission for 24 months post treatment.
Case 3
A three-year-old boy, presented with left facial swelling and left eye proptosis of a few weeks, associated with irritability and difficulty in sleeping. Clinical examination showed left eye proptosis and left periorbital and facial swelling. Nasal flexible endoscopy showed a large purple mass in the left nasal cavity. Imaging CT and MRI showed a large soft tissue mass at the left orbital region, extending to the maxillary sinus, suprasellar region, middle cranial fossa and left masticator space involvement. The mass lesion showed intermediate signal intensity on T1, T2WI, and almost heterogeneous enhancement and appeared restricted on DWI. The patient underwent urgent endoscopic nasal examination under general anesthesia and biopsy from the left sinonasal mass. Pathology confirmed small round malignant cells representing embryonal rhabdomyosarcoma stage IV. The patient received six cycles of chemotherapy in the form of cisplatin and daunorubicin.
Case 4
A six-year-old boy had been complaining of left eye proptosis for a month prior to presentation. Physical examination showed a left bluish large nasal mass obstructing the nasal cavity. Ophthalmological evaluation confirmed the presence of visual loss in the left eye and intact vision in the right eye. Computed tomography of the nose and the paranasal sinuses revealed left nasopharyngeal soft tissue lesion extending superiorly through the inferior orbital fissure into the posterior aspect of the orbit as well as into the anterior aspect of the left cavernous sinus. Endoscopic sinus surgery and debulking of left sinonasal mass were performed. Intraoperative findings showed the origin of the lesion to arise from the left pterygopalatine and infratemporal fossae with a very rich vascular supply from the maxillary artery (Figure 8). Endoscopic medial maxillectomy was performed to access the pterygopalatine fossa and to excise the tumor. Further histopathological analysis revealed small malignant cells, which stained positive with desmin, vimentin, myogenin and WT-1 (cytoplasmic) stains. Negative staining with S-100, synaptophysin, SD99, Pan-CK, and CD45 confirmed the diagnosis of embryonal rhabdomyosarcoma. The patient was referred to pediatric oncology service for staging workup and to start chemoradiation therapy protocols.

Figure 8: Intraoperative endoscopic view of left pterygopalatine and infratemporal fossa lesion (RMS) with increased vascularity from internal maxillary artery.
Discussion
Rhabdomyosarcoma is a rare neoplasm, although it is known to be the most common soft tissue sarcoma among the pediatric population [1, 2]. The term comes from the Greek words, rhabdo (rod shape), myo (muscle), and sarcoma (fleshy growth). This tumor, representing 5-8% of pediatric tumors, considered an aggressive malignant tumor originating from mesenchymal cells of striated muscle, show tendency of early metastasis [3, 4]. RMS was described first in 1854 by Weber. In 1946, Stout described a well-defined morphology of rhabdomyoblast [15]. Several histological subtypes were identified and showed prognostic values. Embryonal (55-60%), alveolar (20-30%), botryoid variant (6%), spindle cell embryonal (3%) and anaplastic (1%) [5]. RMS has been associated with Nevoid basal cell carcinoma (Gorlin syndrome), Neurofibromatosis type 1, Beckwith-Wiedemann syndrome and Rubinstein-Taybi syndrome [6-9]. P53 tumor suppressor gene mutations and rhabdomyosarcoma oncogene mutations have been associated as well [16, 17].
Table 1: The role of surgery in RMS and Ewing’s sarcoma.

Note. Reprinted from "The role of surgery in children with head and neck rhabdomyosarcoma and Ewing's sarcoma" by P.Gradoni, et al. 2010, Surgical Oncology19 (2010) e103-e109, Copyright Ó 2010 Elsevier Inc.
Head and neck has been reported as the most common site of RMS origin (40%). The parameningeal, orbital and non parameningeal RMS approximately represent 50%, 25% and 25% respectively [10, 11]. Parameningeal sites of RMS usually involve the nasopharynx, nasal cavity, paranasal sinuses, middle ear and pterygopalatine fossa. As for the non parameningeal sites, parotid, oral cavity, oropharynx and larynx have been reported in the literature [18]. Orbital and superficial head and neck sites of RMS are considered as more favorable locations of disease compared to parameningeal sites [19, 20].
Pediatric sinonasal rhabdomyosarcoma may initially present with symptoms mimicking rhinosinusitis. Nasal obstruction, rhinorrhea, epistaxis, and headache might subsequently be manifested with further ocular and cranial symptoms [21]. Comprehensive clinical evaluation is crucial; a detailed clinical history and complete physical examination should be obtained. The endoscopic examination that shows a unilateral nasal mass necessitates radiological evaluation with computed tomography, and MRI if indicated, to exclude any vascular lesion or meningioencephalocele. Biopsy and histological evaluation is the most essential step to exclude malignancy, and it can be performed as an office-based endoscopic biopsy or in the operative theater under general anesthesia for uncooperative patients [22].
The majority of pediatric rhabdomyosarcoma demonstrate embryonal and alveolar histological subtypes. The embryonal RMS has been associated with loss of homozygosity on chromosome 11p15 [12]. The alveolar RMS demonstrates a chromosomal translocation [t(2;13)(q35;q14)] which lead to PAX3 gene and FKHR gene fusion and eventually tumor formation [23]. RMS histological examination usually demonstrates small round cells with high cytological variability. Immunohistochemical staining is utilized to confirm the histopathological diagnosis. RMS stained positive with desmin, myogenin and MyoD1. Negative staining with cytokeratin, S-100 and epithelial membrane antigen helps to exclude the other differential diagnoses for RMS [13]. Variable strength of staining to myogenin have been demonstrated between alveolar RMS (strong staining) and embryonal RMS (weak staining) [13, 24].
Intergroup Rhabdomyosarcoma Studies (IRS) established a staging system based on tumor extension and resectability. IRS trials showed improvement in survival with complete tumor resection; however, due to major morbidity after such extensive surgeries and with the development of chemotherapy and radiation therapy, surgical excision is less likely to be done [25-27]. Gatta G et al.’s review of rhabdomyosarcoma results in approximately 67% 5-years survival with multimodality therapy [28]. Rhabdomyosarcoma event-free survival (EFS) been reported lately were incentive [29, 30]. Ma X, et al. retrospective multicenter study results in 10 years EFS of 53.4 ± 5.1% [31]. Singapore rhabdomyosarcoma experience reported by Aung L et al. optimistically reported 5-years EFS: of 75% [32].
The role of surgery in RMS may be limited for obtaining biopsies or for palliative purposes [14]. A few cases of nonparameningeal and non-orbital can be resected without major morbidities, in contrast to parameningeal tumors [33]. Only a few studies focused on the effectiveness and application of surgical resection [22]. P.Gradoni et al. reviewed and highlighted the role of surgery in children with head and neck rhabdomyosarcoma and Ewing’s sarcoma. Excluding orbital RMS, the review explained the surgical role including biopsy, debulking surgery, radical surgery as primary treatment modality, surgery after primary chemoradiation, neck dissection, salvage surgery after locoregional relapse and metastasectomy (Table 1) [34].
Popular antineoplastic agents used to treat RMS include vincristine, cyclophosphamide, actinomycin D, and Adriamycin [25-27]. These agents helped to improve the cure rate from 25% to 75% during the past 40 years. Unfortunately, parameningeal RMS is difficult to treat; reports showed more than forty percent of cases relapsed. Chemotherapy with alkylating agents have achieved a relapsed free survival 90% for nonparameningeal tumors and 65% for parameningeal tumors [35].
Conclusion
Pediatric sinonasal malignancies, including rhabdomyosarcoma may initially present with symptoms similar to rhinosinusitis, causing a delay in proper referral, diagnosis and management. Careful inspection and comprehensive examination with nasal endoscopy is crucial. Rhabdomyosarcoma incorporative management consists of radiation therapy and chemotherapy. The surgeon has a major role in providing the specimen for histopathological diagnosis. This article highlights the nature of pediatric RMS and emphasizes the surgeon to have low threshold in evaluating pediatric sinonasal masses to avoid diagnosis and management delay in order to increase survival and cure rates. Surgical role in RMS may further be investigated in order to obtain standardized criteria for surgical interventions.
Conflicts of Interest
None.
Article Info
Article Type
Case StudyPublication history
Received: Mon 02, Mar 2020Accepted: Wed 18, Mar 2020
Published: Fri 20, Mar 2020
Copyright
© 2023 Ali Al Momen . This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited. Hosting by Science Repository.DOI: 10.31487/j.IJSCR.2020.01.08
Figures & Tables
Table 1: The role of surgery in RMS and Ewing’s sarcoma.
Note. Reprinted from "The role of surgery in children with head and neck rhabdomyosarcoma and Ewing's sarcoma" by P.Gradoni, et al. 2010, Surgical Oncology19 (2010) e103-e109, Copyright Ó 2010 Elsevier Inc.
References
- Maurer HM (1984) Clinical pediatric oncology. St Louis, MO Mosby Year B.
- Arndt CAS, Crist WM (1999) Common musculoskeletal tumors of childhood and adolescence. N Engl J Med 341: 342-352. [Crossref]
- Crist WM, Kun LE (1991) Common solid tumors of childhood. N Engl J Med 324: 461-471. [Crossref]
- McDowell HP (2003) Update on childhood rhabdomyosarcoma. Arch Dis Child 88: 354-357. [Crossref]
- Duan F, Smith LM, Gustafson DM, Zhang C, Dunlevy MJ et al. (2012) Genomic and clinical analysis of fusion gene amplification in rhabdomyosarcoma: a report from the Children’s Oncology Group. Genes Chromosomes Cancer 51: 662-674. [Crossref]
- McKeen EA, Bodurtha J, Meadows AT, Douglass EC, Mulvihill JJ (1978) Rhabdomyosarcoma complicating multiple neurofibromatosis. J Pediatr 93: 992-993. [Crossref]
- Singer DB (1991) Textbook of Fetal and Perinatal Pathology, Vol 1. Blackwell Science Incorporated.
- Beddis IR, Mott MG, Bullimore J (1983) Case report: nasopharyngeal rhabdomyosarcoma and Gorlin’s naevoid basal cell carcinoma syndrome. Med Pediatr Oncol 11: 178-179. [Crossref]
- Ruymann FB, Maddux HR, Ragab A, Soule EH, Palmer N et al. (1988) Congenital anomalies associated with rhabdomyosarcoma: an autopsy study of 115 cases. A report from the Intergroup Rhabdomyosarcoma Study Committee (representing the Children's Cancer Study Group, the Pediatric Oncology Group, the United Kingdom Children's Cancer Study Group, and the Pediatric Intergroup Statistical Center). Med Pediatr Oncol 16: 33-39. [Crossref]
- Hicks J, Flaitz C (2002) Rhabdomyosarcoma of the head and neck in children. Oral Oncol 38: 450-459. [Crossref]
- Months SR, Raney RB (1986) Rhabdomyosarcoma of the head and neck in children: the experience at the Children’s Hospital of Philadelphia. Med Pediatr Oncol 14: 288-292. [Crossref]
- El Badry OM, Minniti C, Kohn EC, Houghton PJ, Daughaday WH et al. (1990) Insulin-like growth factor II acts as an autocrine growth and motility factor in human rhabdomyosarcoma tumors. Cell Growth Differ 1: 325-331. [Crossref]
- Kumar S, Perlman E, Harris CA, Raffeld M, Tsokos M (2000) Myogenin is a specific marker for rhabdomyosarcoma: an immunohistochemical study in paraffin-embedded tissues. Mod Pathol 13: 988-993. [Crossref]
- Healy GB, Upton J, Black PM, Ferraro N (1991) The role of surgery in rhabdomyosarcoma of the head and neck in children. Arch Otolaryngol Head Neck Surg 117: 1185-1188. [Crossref]
- Stout AP (1946) Rhabdomyosarcoma of the skeletal muscles. Ann Surg 123: 447-472. [Crossref]
- Stratton MR, Fisher C, Gusterson BA, Cooper CS (1989) Detection of point mutations in N-ras and K-ras genes of human embryonal rhabdomyosarcomas using oligonucleotide probes and the polymerase chain reaction. Cancer Res 49: 6324-6327. [Crossref]
- Felix CA, Kappel CC, Mitsudomi T, Nau MM, Tsokos M et al. (1992) Frequency and diversity of p53 mutations in childhood rhabdomyosarcoma. Cancer Res 52: 2243-2247. [Crossref]
- Miloglu O, Altas SS, Buyukkurt MC, Erdemci B, Altun O (2011) Rhabdomyosarcoma of the oral cavity: a case report. Eur J Dent 5: 340-343. [Crossref]
- Malempati S, Hawkins DS (2012) Rhabdomyosarcoma: Review of the Children’s Oncology Group (COG) Soft-Tissue Sarcoma Committee Experience and Rationale for Current COG Studies. Pediatr Blood Cancer 59: 5-10. [Crossref]
- Womer RB, Barr FG, Linardic CM (2015) Rhabdomyosarcoma. Nathan Oski’s Hematol Oncol Infancy Child 1906-1945.e10.
- Herrmann BW, Sotelo Avila C, Eisenbeis JF (2003) Pediatric sinonasal rhabdomyosarcoma: three cases and a review of the literature. Am J Otolaryngol 24: 174-180. [Crossref]
- Fyrmpas G, Wurm J, Athanassiadou F, Papageorgiou T, Beck JD et al. (2009) Management of paediatric sinonasal rhabdomyosarcoma. J Laryngol Otol 123: 990-996. [Crossref]
- Shapiro DN, Sublett JE, Li B, Downing JR, Naeve CW (1993) Fusion of PAX3 to a member of the forkhead family of transcription factors in human alveolar rhabdomyosarcoma. Cancer Res 53: 5108-5112. [Crossref]
- Cessna MH, Zhou H, Perkins SL, Tripp SR, Layfield L et al. (2001) Are myogenin and myoD1 expression specific for rhabdomyosarcoma? A study of 150 cases, with emphasis on spindle cell mimics. Am J Surg Pathol 25: 1150-1157. [Crossref]
- Maurer HM, Beltangady M, Gehan EA, Crist W, Hammond D et al. (1988) The Intergroup Rhabdomyosarcoma Study-I. A final report. Cancer 61: 209-220. [Crossref]
- Maurer HM, Gehan EA, Beltangady M, Crist W, Dickman PS et al. (1993) The Intergroup Rhabdomyosarcoma Study-II. Cancer 71: 1904-1922. [Crossref]
- Crist W, Gehan EA, Ragab AH, Dickman PS, Donaldson SS et al. (1995) The Third Intergroup Rhabdomyosarcoma Study. J Clin Oncol 13: 610-630. [Crossref]
- Gatta G, Botta L, Rossi S, Aareleid T, Bielska Lasota M et al. (2014) Childhood cancer survival in Europe 1999-2007: results of EUROCARE-5--a population-based study. Lancet Oncol 15: 35-47. [Crossref]
- Salman M, Tamim H, Medlej F, El Ariss T, Saad F et al. (2012) Rhabdomyosarcoma Treatment and Outcome at a Multidisciplinary Pediatric Cancer Center in Lebanon. Pediatr Hematol Oncol 29: 322- 334. [Crossref]
- Park JA, Kim EK, Kang HJ, Shin HY, Kim IH et al. (2008) Initial response to treatment was highly associated with the prognosis of childhood rhabdomyosarcoma: a retrospective analysis of a single center experience in Korea. Cancer Res Treat 40: 111-115. [Crossref]
- Ma X, Huang D, Zhao W, Sun L, Xiong H et al. (2015) Clinical characteristics and prognosis of childhood rhabdomyosarcoma: a ten-year retrospective multicenter study. Int J Clin Exp Med 8: 17196-17205. [Crossref]
- Aung L, Soe TA, Chang KT, Quah TC (2014) Singapore rhabdomyosarcoma (RMS) experience: shall we change our practice. Ann Acad Med Singapore 43: 86-95. [Crossref]
- Daya H, Chan HS, Sirkin W, Forte V (2000) Pediatric rhabdomyosarcoma of the head and neck: is there a place for surgical management? Arch Otolaryngol Head Neck Surg 126: 468-472. [Crossref]
- Gradoni P, Giordano D, Oretti G, Fantoni M, Ferri T (2010) The role of surgery in children with head and neck rhabdomyosarcoma and Ewing’s sarcoma. Surg Oncol 19: e103-e109. [Crossref]
- Baker KS, Anderson JR, Link MP, Grier HE, Qualman SJ et al. (2000) Benefit of intensified therapy for patients with local or regional embryonal rhabdomyosarcoma: results from the Intergroup Rhabdomyosarcoma Study IV. J Clin Oncol 18: 2427-2434. [Crossref]