Journals
Is it all just an Akt you’d be SMAD to believe it! Role of TGFβ1 in oral cancer metastasis
A B S T R A C T
Introduction: TGFβ1 activates both SMAD and non-SMAD dependent signalling pathways but their role in oral cancer cell migration and metastasis is little known. The aim of this research was to investigate the role of TGFβ1-induced signalling pathways in oral cancer cell migration and to establish whether the inhibition of the associated pathway may be a suitable target for chemotherapeutic drug design to control oral cancer cell metastasis.
Materials and Methods: SDS-PAGE and Western blot techniques were used to investigate the expression and phosphorylation status of TGFβ1- induced key signalling molecules such as, SMAD, Akt and MAPK in normal keratinocytes and oral cancer cells. Gap closure and scatter assays were employed to study the effect of TGFβ1 on cell migration. Akt and MAPK inhibitors were also used to explore the role of associated signalling pathways in TGFβ1-induced cell migration. Phosphorylation of Akt, MAPK and SMAD were analysed by immunofluorescence in TGFβ1-induced migrated cells.
Results: TGFβ1 stimulated the phosphorylation of SMAD, Akt and MAPK in both normal keratinocytes and oral cancer cells. The level of phosphorylation, however, depended upon cell type, time of exposure to and concentration of TGFβ1. TGFβ1-stimulated cancer cell migration both as single cells (EMT, epithelial to mesenchymal transition) and as a sheet of cells. Inhibitor assays confirmed that cancer cell migration is phosphorylated-Akt dependent. However, TGFβ1 did not stimulate migration as a sheet of cells, but EMT of normal keratinocytes which was phosphorylated-MAPK dependent.
Conclusion: TGFβ1-induced oral cancer cell migration was dependent on the Akt signalling pathway. From this study, we propose that blocking the Akt pathway may inhibit oral cancer metastasis and could have potential in translating this research into clinical practice.
Keywords
TGFβ1, Akt, SMAD, MAPK, oral cancer, cell migration, cell signalling
Abbreviations
TGFβ1-Transforming Growth Factor beta 1, MAPK-Mitogen Activated Protein Kinase, EMT-Epithelial to Mesenchymal Transition, pAkt T308- Phosphorylated Akt at Threonine 308, pAkt S473-Phosphorylated Akt at Serine 473, OSCC- Oral Squamous Cell Carcinoma HNSCC- Head and Neck Squamous Cell Carcinoma
I N T R O D U C T I O N
Head and neck cancer are the sixth most common cancer in the world and squamous cell carcinoma is the most predominant. Oral cancer is a type of head and neck cancer which includes cancers of the lips, tongue, cheeks, floor of the mouth, hard and soft palate, sinuses, and pharynx (throat). While oral cancer patients have a 5-year survival rate below 40%, patients with metastatic disease have an extremely poor prognosis and a survival rate lower than 10% [1, 2].The development of distant metastases, second primary tumours and potentially inoperable recurrences with increased resistance to radiation or chemotherapy are the main reasons for mortality [3]. Cell motility or migration is an essential part of most tumour metastatic and wound healing pathways. Cells need to migrate away from their microenvironment to enable the tumour to spread or metastasise. Cell migration can be divided into collective (migration as a group/sheet of cells without completely disrupting their cell-cell contacts) and single cell migration [4, 5]. Most of the single cell migration is accomplished by epithelial to mesenchymal transition (EMT) which is an important biological phenomenon characterised by changes in morphology from cuboidal epithelial to spindle shape mesenchymal, overexpression of vimentin (a mesenchymal marker) and loss of E-cadherin (an epithelial marker) [6]
Earlier evidence suggested that, growth factors and matrix macromolecules are essential for the movement of cells [7]. Previous studies in our laboratories showed that Vascular Endothelial Growth Factor (VEGF), Epidermal Growth Factor (EGF) and Transforming Growth Factor alpha (TGFα) stimulate both fibroblast and oral cancer cells to migrate. We have previously shown that the PI3 kinase/Akt pathways are essential for the migration of fibroblasts and oral cancer cells in response to added factors. The addition of PI3 kinase/Akt inhibitors block the migration stimulating activity of VEGF, EGF and TGFα, the data indicating that those growth factors increase phosphorylation of Akt at Thr 308 and Ser 473 [8-10]. The role of TGFβ1, however, is still unknown in oral cancer cell metastasis. In contrast to its tumour suppressive effect, TGFβ signalling also promotes tumour progression by means of loss of epithelial cell adhesion, extracellular matrix remodelling and enhanced angiogenesis [11]. A study using cancer tissues reported that TGFβ1 was overexpressed in 36% of oesophageal cancer [12]. Pasini et al (2001) also reported increased levels of TGFβ1 mRNA levels in HNSCC [13]. Interestingly, the percentage of TGFβ1 overexpression in oral cancer being the highest (66%) among tumours from different sites of HNSCC [14]. Approximately 78% of HNSCC samples also exhibited a 1.5- to 7.5-fold increase in TGFβ in comparison with normal controls [14]. TGF-β initiates Smad as well as non-Smad dependent signalling, such as PI3K/Akt, P44/42 MAPK (Erk1/2), p38 MAPK, and Rho-GTPase pathways [15, 16]. TGF-β acts as a common and potent inducer of EMT by combining both Smad and non-Smad signalling pathways [17-19].
In this study we aimed to investigate the role of downstream signalling pathways in TGFβ1-induced oral cancer cell migration. The resultant data increases the spectrum of knowledge of the TGFβ induced pathways and to propose that inhibition of pathway would be a suitable target for chemotherapeutic drug design to control oral cancer cell metastases.
Materials and Methods
I Reagents, antibodies and inhibitors
The primary antibodies used were: rabbit monoclonal anti-pAkt T308 (# 2965), anti-pAkt S473 (# 4060), anti-pan Akt (# 4691), anti-SMAD2/3 (#8685), anti-p-SMAD2/3 (# 8828), anti-P44/42 MAPK (# 9102), anti- phospho P44/42 MAPK (#4370), anti- E-cadherin (# 3195) and anti-Vimentin (# 3932) (all Cell Signaling Technology, Inc., Danvers, MA, USA). The secondary antibodies used were goat anti-rabbit HRP conjugated (# 7074, Cell Signaling Technology), and goat anti-rabbit alexa fluor 488 conjugated (# 4412, Cell Signaling Technology). Recombinant Human TGFβ1 (# 77039) was purchased from Sigma, St. Lous, MO, USA. The PI3K-Akt pathway inhibitors MK2206 (# S1078) was purchased from Seleckchem, Houston, USA and MAPK inhibitor, PD98059 (#9900) was purchased from Cell Signaling Technology.
II Cell culture
The oral squamous cell carcinoma (OSCC) cell line (TYS), derived from floor of the mouth was a kind gift from Dr. Koji Harada, University of Tokushima, Japan. Normal adult keratinocytes (HaCaT) were a kind gift from Professor S.L. Schor (late), Dundee Dental School, UK. All cells were cultured at 37°C and 5% CO2 in minimum essential medium (MEM) supplemented with 10% (v/v) heat-inactivated foetal calf serum and 200 mM glutamine.
III Cell lysis, SDS-PAGE and Western blot
60-70 % confluent cells were treated with the experimental conditions (TGFβ1 ± inhibitors) for a maximum 48 hours and serum starved overnight. Cells were then lysed on ice with RIPA buffer (50 mM Tris HCl, 150 mM NaCl, pH 7.4; 0.1% w/v SDS, 1% v/v Triton x-100, 1% w/v sodium deoxycholate and 5 mM EDTA) containing protease inhibitors (Roche Applied Science, IN, USA) at different time points. Lysates were clarified by centrifugation at 13,000 rpm for 5 minutes. Samples were then mixed with an equal volume of Laemmli sample loading buffer (BioRAD, CA, USA) including 5% (v/v) 2-mercaptoethanol. Samples were heated at 95 °C for 5 minutes and loaded onto either 10% or ‘Any kD’ SDS-PAGE BioRad stain-free precast gels. After completion of SDS PAGE, gel images were captured using Gel Doc imaging system (BioRad) for total protein and were then electro-transferred onto PVDF transfer membrane (BioRad). Blots were then immunolabelled with anti-pAkt T308 (1:1000), anti-pAkt S473 (1:2000), anti-pan Akt (1:1000), anti- SMAD2/3(1:1000), anti-pSMAD 2/3 (1:1500), anti-P44/42 MAPK (ERK1/2) (1:1000) and anti-phospho P44/42 MAPK (pERK1/2) (1:2000) and goat anti-rabbit HRP conjugated secondary antibody (1:2000). Immunoblots were developed using Immun-Star WesternC Kit (BioRad) and images were captured using a Gel Doc imaging system. Total protein gel images were used to normalise and quantify the respective blot by Image lab software (BioRad) instead of housekeeping gene such as GAPDH. Images were then cropped and processed using ImageJ software (NIH, Bathesda, USA).
IV Scatter assay and immunofluorescent staining
Cell scattering is a dynamic process for investigating EMT in which dispersion of compact colonies of epithelial cells is induced by certain soluble factors such as growth factors [20]. The assay was performed as described earlier [10]. In brief, cells were seeded at a density of 2 × 104 cells/ml, in 60 mm dishes and grown until small well-defined colonies were visible. The cells were washed twice, serum starved overnight, and incubated in test conditions for 48 hours. Scattering of the cells was observed by at least two individuals before images were taken. Images were captured using SC50 digital camera attached to inverted microscope (IX70, Olympus, Tokyo, Japan) at either 100X or 200X magnification.
After 48 hours, the cells were fixed with cold methanol for 15 minutes and then washed with phosphate-buffered saline (PBS). Cells were then treated with 0.2% (v/v) Triton X-100 in PBS for 5 minutes, small areas of the dishes were ringed with Immunopen (DAKO, Cambridgeshire, UK) and blocked with 5% (v/v) normal goat serum (NGS; Vector Laboratories, Burlingame, CA, USA) in PBS with 0.1% (v/v) Tween 20 (PBST) for 30 minutes. The cells were then incubated with anti-E-cadherin (1:200), anti-vimentin (1:50), anti-pAkt T308 (1:1600 #13038, Cell Signaling Technology), anti-pAkt S473(1:200), anti-P44/42 MAPK (1:100), anti- phospho P44/42 MAPK (1:200) rabbit monoclonal primary antibodies diluted in 5% (v/v) NGS in PBST and kept at 4°C for overnight. They were then washed twice with PBST, once with PBS and incubated with anti-rabbit immunoglobulin G (H+L), F(ab′)2 Fragment (Alexa Fluor® 488 Conjugate) (1:1000) for 1 hour at room temperature. After washing twice with PBST and once with PBS, sections were cover slipped with aqueous mounting medium (Sigma-Aldrich, St Louis, MO, USA). Sections were then viewed with an Olympus IX70 inverted fluorescent microscope (Olympus, Tokyo, Japan) at 200X or 400X magnification. Images were captured using a XM10 digital camera (Olympus). All devices were controlled, and images were processed by CellSense software (Olympus).
V Scratch assay
The scratch assay, a directional in vitro 2D migration assay, was performed as described earlier [21]. A cell monolayer was serum starved overnight and then a wound was made in the monolayer using a 100μl pipette tip. The cells were then incubated in test conditions (TGFβ1 ± MK2206) for 24 hours. Images were captured using an IX70 inverted microscope start at t=0 and at the end point to monitor cell migration causing wound closure. Scratch images were then quantified using ImageJ macros-MRI wound healing tool.
VI Modified gap closure assay
A cell culture inserts (#81176, IBIDI GmbH, Munich, Germany) was used to modify the traditional gap closure/wound healing/scratch assay. Instead of making a scratch in a cell monolayer where cells are wounded, a culture insert was used to create a cell-free gap. Two cell culture reservoirs in the insert allowed cell growth in the designated areas after filling them with the cell suspension. The culture insert was removed after 24 hours of cell attachment which created a cell-free gap of approximately 500 µm. Test conditions (TGFβ1 ± MK2206) were then applied and incubated for 24 hours. Images were captured using an inverted microscope at the start and end of the experiment at 100X magnification and quantified using ImageJ macros-MRI wound healing tool.
VII Statistical Analysis
The data was analysed by Graphpad Prism (v5.0) (La Jolla, CA, USA). All the experiments were repeated at least three times. Differences between groups were evaluated with the Student's t-test. Differences were considered significant when the p value was less than 0.05.
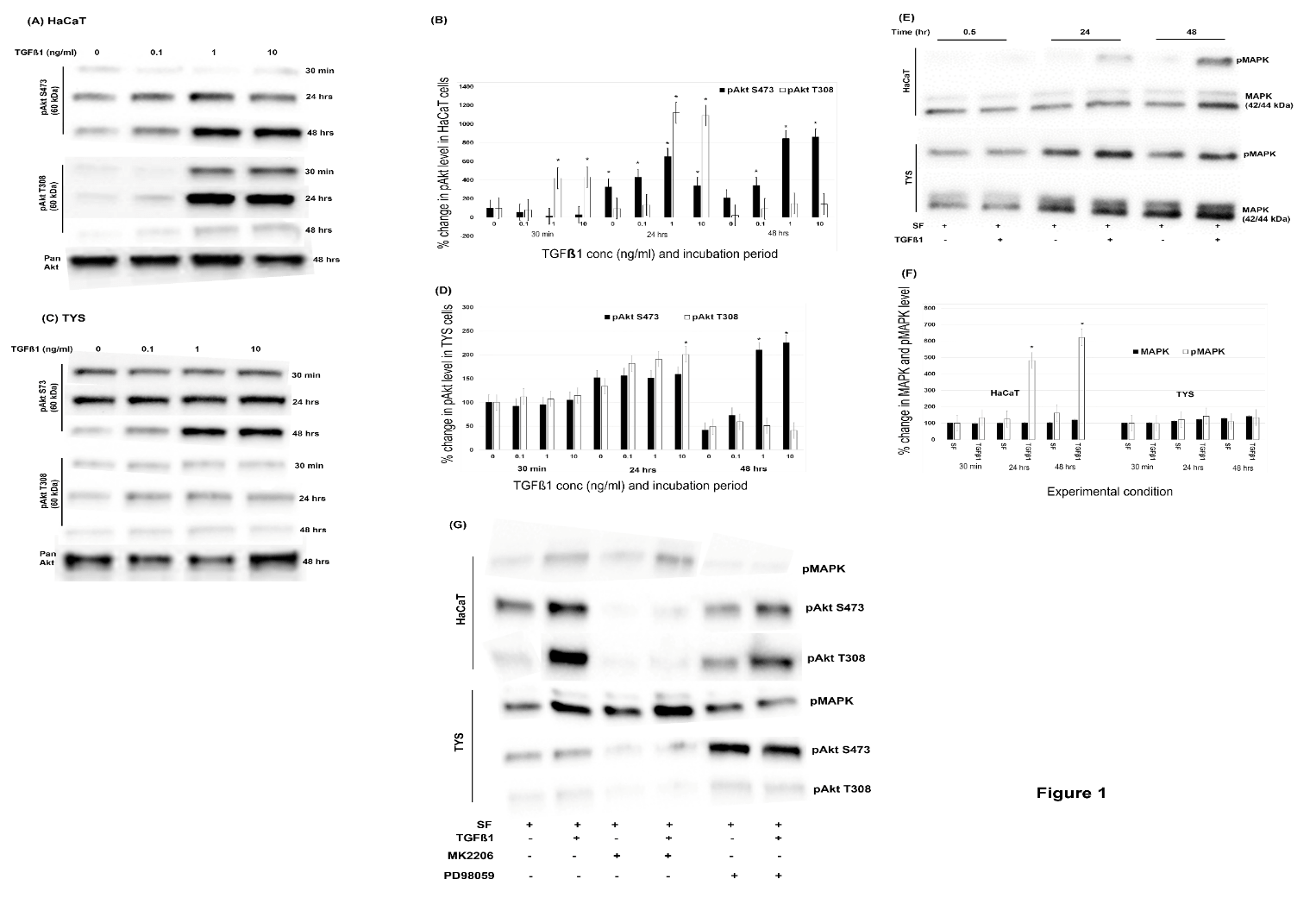
Figure 1: (TGFβ1 stimulated non-SMAD pathways which was time and concentration dependent. (A) Normal keratinocytes and (C) oral cancer cells were incubated with different concentrations of TGFβ1 for a maximum of 48 hours and lysed at different time points. Lysates were then analysed by SDS-APGE and WB experiments for pAkt T308, pAkt S473 and pan Akt antibodies. (B) Percent changes in pAkt level in HaCaT cells and (D) in oral cancer cells. Blot images were normalised against total protein and quantified using ImageLab software. All the experimental conditions were compared with the control (SF-serum free medium at 30 minutes). (E) P44/42 MAPK and phospho-P44/42 MAPK antibodies were also applied in 10 ng/ml TGFβ1 treated cell lysates. Bands were observed at 42 and 44 kDa. (F) Quantification of the level of changes in total MAPK and phosphorylated MAPK in response to TGFβ1 at different times (G) Cells were treated with 5µM MK2206 and 50µM PD98059 with or without 10ng/ml TGFβ1 for 24 hours. Cell lysates were then analysed by SDS-APGE and WB experiments for p-MAPK, pAkt S473 and pAkt T308 antibodies. All the images were cropped from the original blot images. SF (serum-free MEM medium) used as a negative control. (*) denotes to p<0.05.
Results
I TGFβ1 triggered both SMAD and non-SMAD dependent pathways which was time dependent
Normal adult keratinocytes and oral cancer cells were treated with different concentrations TGFβ1 for 48 hours and both cells phosphorylated Akt at Ser473 and Thr 308 residues, but the level of phosphorylation was different. TGFβ1 (10 ng/ml) increased the phosphorylation of Akt at S473 to a level approximately 9-fold in HaCaT cells (p < 0.05) (Figure 1A, 1B) and more than 2-fold in TYS cells than the control after 48 hours (p < 0.05) (Figure 1C, 1D). TGFβ1 also increased the phosphorylation of Akt at T308 approximately 11-fold in HaCaT (p < 0.05) (Figure 1A, 1B) and 2-fold in TYS cells than the control after 24 hours (p < 0.05) and the level decreased to lower than control (TYS) (Figure 1C, 1D) and back to control levels (HaCaT) after 48 hours (Figure 1A,1B). The amount of Pan Akt was consistent in both cell lines treated with different concentrations of TGFβ1 for 48 hours (Figure 1A, 1C). TGFβ1 also increased the phosphorylation of MAPK in HaCaT cells about 6-fold and about 1.5-fold in TYS than that of control after 48 hours (Figure 1E, 1F). The level of MAPK expression was consistent in both cell lines (Figure 1E, 1F). The Akt inhibitor (MK2206) and MAPK inhibitor (PD98059) effectively blocked the phosphorylation of Akt and MAPK respectively, in normal keratinocytes. MK2206 effectively blocked TGFβ1-induced Akt phosphorylation in oral cancer cells but PD98059 blocked the phosphorylation of MAPK only in 44 kDa isoform or ERK1 (Figure 1G). Cells treated with 10 ng/ml TGFβ1 for 24 hours showed that total SMAD2/3 expression was not changed compared to that of control. TGFβ1 also stimulated the phosphorylation of SMAD2/3 5 times higher in HaCaT (p < 0.05) and more than 2 times higher in TYS than the control after 2 hours (p < 0.05) and the level of phosphorylation went down after 4 hours in both cell lines (Figure 5A, 5B).
II TGFβ1 stimulated the scattering of both cell lines via two separate signalling pathways.
Small colonies of normal adult keratinocytes were treated with 10 ng/ml TGFβ1 for 48 hours and cells started to scatter out from these colonies after 24 hours. After 48 hours, a mesenchymal-type cell morphology was observed in the scattered cells. MAPK inhibitor, PD98059, effectively blocked the TGFβ1-induced scattering of normal keratinocytes, whereas the Akt inhibitor, MK2206, did not block scattering (Figure 2A). Oral cancer cells also scattered from colonies as mesenchymal-type cells after 24 hours and this morphology persisted for 48 hours. Scattering of oral cancer cells, however, was effectively blocked by the Akt inhibitor, MK2206. MAPK inhibitor, PD98059 could not block the TGFβ1-induced scattering of oral cancer cells (Figure 2B).

Figure 2: (A) TGFβ1-induced normal keratinocyte (HaCaT) scattering/migration is MAPK dependent and (B) oral cancer (TYS) scattering/migration is Akt dependent. Cells were treated with 10 ng/ml TGFβ1 alone and/or in combination with 5µM MK2206 and 50µM PD98059 for 48 hours. Cells scattering was observed for 48 hours and images were captured at different time points. An inverted microscope with a digital camera was used to capture the images at either 100x or 200X magnification. Cells were fixed after 24 hours and 48 hours of observation. SF (serum-free MEM medium) was used as a negative control.
III TGFβ1-induced oral cancer cells scattered through EMT after 24 hours with strong Akt and MAPK and no SMAD 2/3 phosphorylation
Cells treated with TGFβ1 for 48 hours showed high levels of vimentin expression and low levels of E-cadherin expression, which corroborated the idea that the cells scattered as a result of EMT. Serum-free MEM treated non-scattered cells showed strong E-cadherin expression at cell-cell junctions (Figure 3). Phosphorylation of Akt at Ser473 was higher in TGFβ1-induced scattered normal keratinocytes (Figure 3) after 24 hours compared to the control and pAkt T308. Phosphorylated Akt in both residues, however, was stronger in the oral cancer scattered cells than the serum-free medium treated non-scattered cells (Figure 4). Phosphorylation of MAPK, however, was higher in TGFβ1-induced scattered oral cancer cells (highest at 24 hours) and in normal keratinocytes (highest at 48 hours) compared to cells treated with serum-free MEM medium (control) (Figure 3 & 4). No phosphorylation of SMAD2/3 was observed in TGFβ1-induced scattered oral cancer cells after 24 hours, however, a low level of phosphorylation of SMAD2/3 was observed in scattered normal keratinocytes after 24 hours. A low-level phosphorylation of SMAD2/3 was observed after 2 hours of TGFβ1 treatment in oral cancer cells compared to the control (Figure 5C).

Figure 3: TGFβ1-induced normal keratinocytes (HaCaT) scattered through EMT. Phosphorylation of both Akt S473, MAPK and vimentin expression were visualised by Immunofluorescence using anti-E-cadherin, anti-vimentin, anti-pAkt S473, anti-pAkt T308 and anti-pMAPK antibodies after 48 hours of scattering. Images were captured by an inverted fluorescence microscope and CellSense software either at 200x or 400X magnification and processed by ImageJ software. SF (serum-free MEM medium) was used as a negative control.
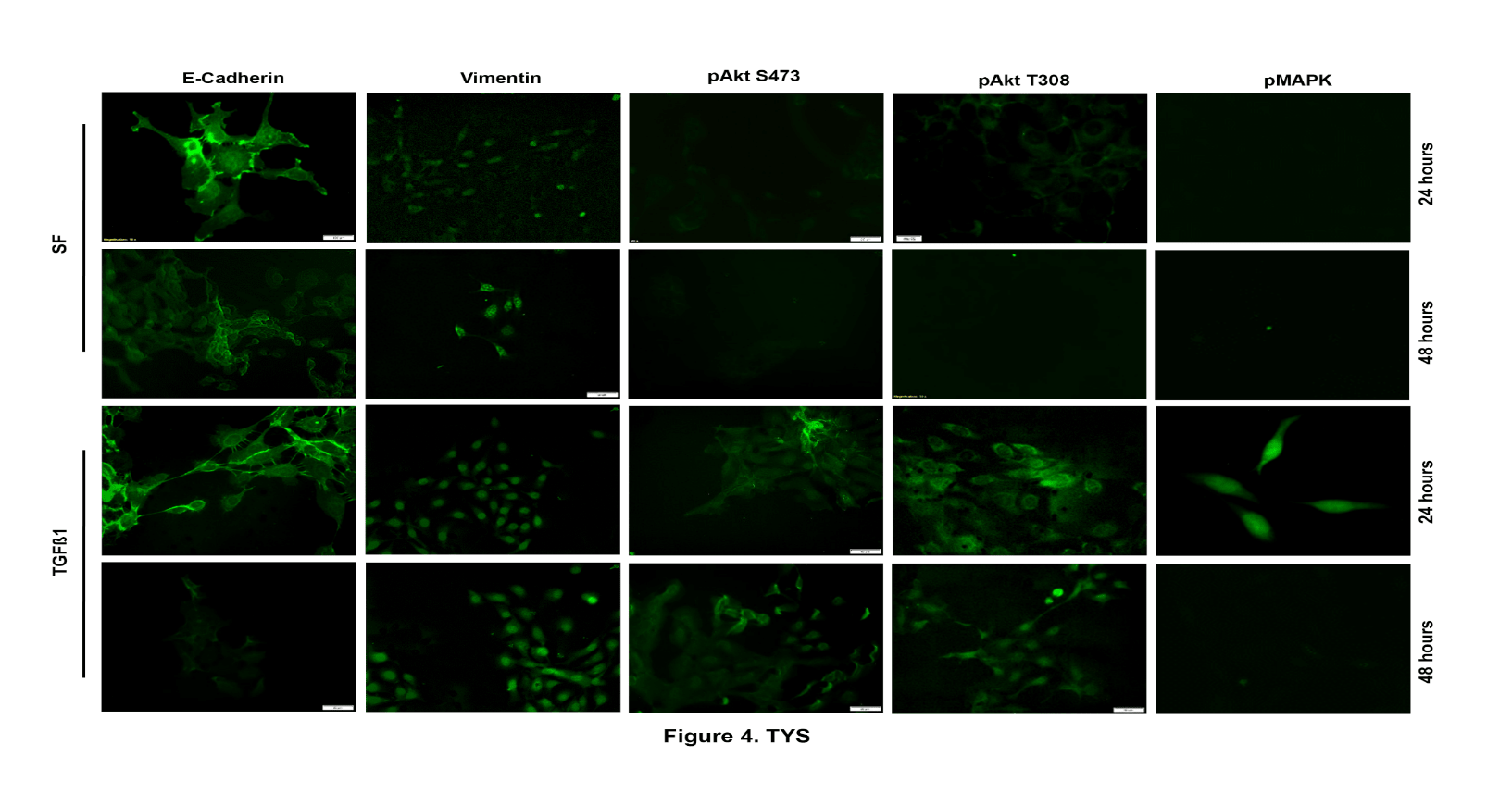
Figure 4: TGFβ1-induced oral cancer cell (TYS) scattered through EMT and higher pAkt was observed in scattered cells than the control after 48 hours. Fixed scattered cells were labelled with anti-E-cadherin, anti-vimentin, anti-pAkt S473, anti-pAkt T308 and anti-pMAPK antibodies and visualised by an Alexa Fluor ® tagged secondary antibody. Images were captured by inverted fluorescence microscope and CellSense software. Images were captured either at 200x or 400X magnification and processed by ImageJ software. SF (serum-free MEM medium) was used as a negative control.
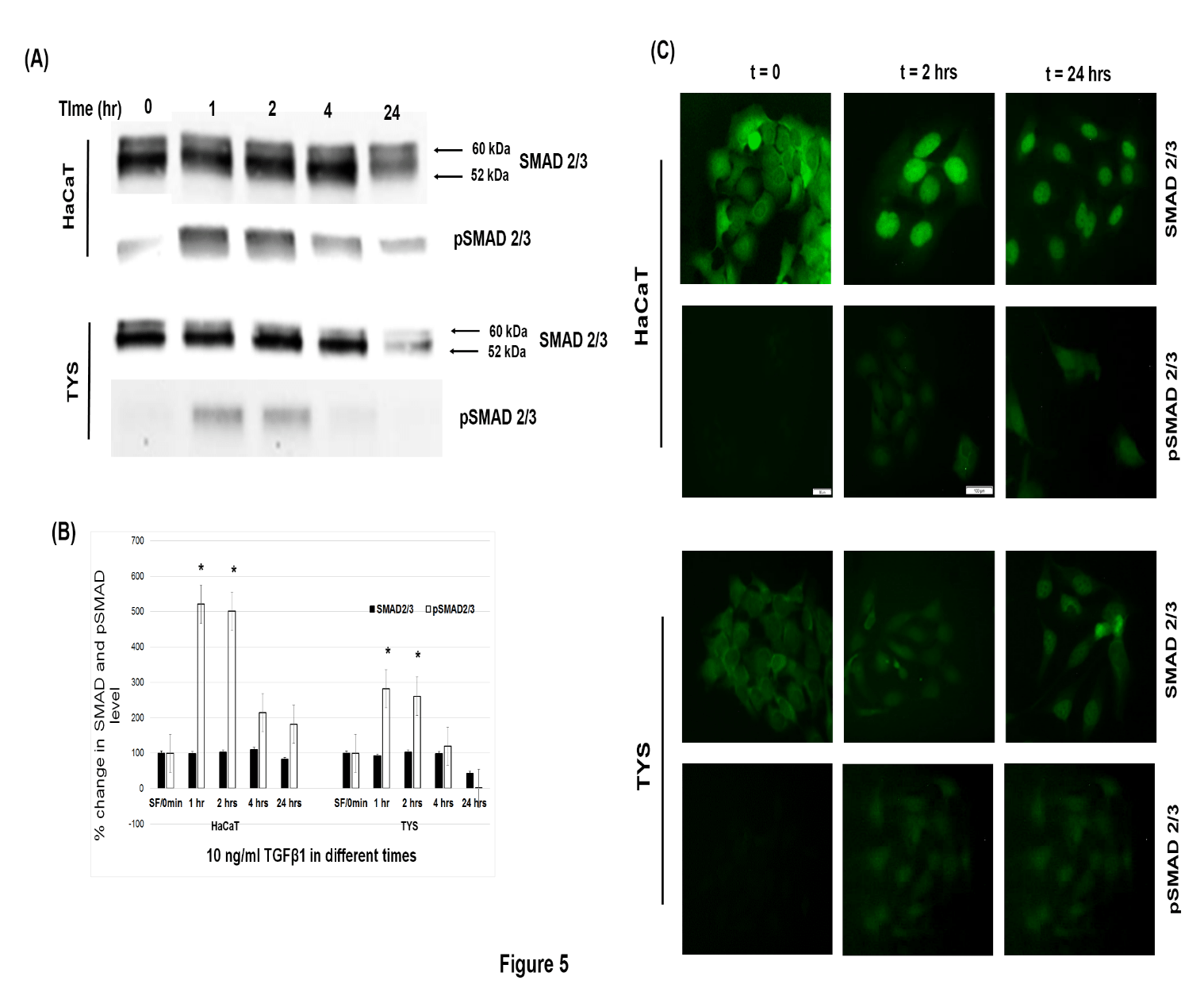
Figure 5: SMAD phosphorylation was persisted for 2 hours in oral cancer. (A) Cells were incubated by 10 ng/ml TGFβ1 for 24 hours and lysed at different time points. SMAD 2/3 and pSMAD2/3 antibodies were then used in WB experiments. (B) Quantification of the level of changes in total SMAD and phosphorylated SMAD in response to TGFβ1 at different times. Blot images were normalised against total protein and quantified using ImageLab software. All the experimental conditions were compared with the control (SF-serum free medium). (*) denotes to p < 0.05. (C) Cells were treated with 10 ng/ml TGFβ1 for 24 hours and fixed at different time points. Cells were then labelled with anti-SMAD2/3 and anti-pSMAD2/3 antibodies and visualised by an Alexa Fluor ® tagged secondary antibody. Images were captured by an inverted fluorescence microscope and CellSense software. Images were captured either at 200x or 400X magnification and processed by ImageJ software.
IV TGFβ1-induced oral cancer cell migration is Akt dependent
TGFβ1 stimulated the migration of oral cancer cells in both traditional scratch (Figure 6A) and modified gap closure (Figure 6B) assays. The gap created by either scratch or inserts in the oral cancer cell monolayer was closed completely after 24 hours by cell migration stimulated by 10 ng/ml TGFβ1 (p < 0.05) (Figure 6A, 6B, 6C). Normal keratinocytes, however, were not stimulated to migrate by exogenous TGFβ1 in the scratch assay (Figure 6A). MK2206, the Akt inhibitor, blocked about 60% of TGFβ1-induced cell migration of oral cancer cells (Figure 6A, 6C).

Figure 6: TGFβ1 stimulated oral cancer cell migration in an Akt-dependent manner in the gap closing assay. (A) Cells were treated with 10 ng/ml TGFβ1 with or without Akt inhibitor, MK2206 (5 µM) for 24 hours in the scratch/wound healing assay. (B) Oral cancer cells were treated with 10 ng/ml TGFβ1 with or without the Akt inhibitor, MK2206 (5 µM) for 24 hours in the modified gap closing assay. Images were captured by inverted fluorescence microscope and CellSense software at 100x magnification and processed by ImageJ software. SF (serum-free MEM medium) was used as a negative control. (C) Quantification of modified gap closure assay. Images were quantified by an ImageJ-macros, MRI wound healing tools. All the experimental conditions were compared with the negative control. (*) denotes to p<0.05.
Discussion
Protein expression and status of phosphorylation study confirmed that TGFβ1 can induce both SMAD-dependent and non-SMAD (PI3K, MAPK) dependent signalling pathways. This study showed that TGFβ1 phosphorylates SMAD2/3, Akt and MAPK in both normal keratinocytes and oral cancer cells. The phosphorylation of those signalling proteins in response to TGFβ1, was however time dependent. Zi et al (2012) also reported in their mathematical modelling study that TGFβ1 signalling is time dependent [22]. Phosphorylation of the SMAD2/3 was observed as an early event and only persisted for 2 hours in oral cancer cells, whereas both Akt and MAPK phosphorylation started as early as 30 minutes after stimulation and persisted for 48 hours. Akt phosphorylation at S473 residue was higher than that of T308 residue in the oral cancer cell line. The phosphorylation level of Akt T308 went down after 24 hours, while pAkt S473 levels were highest after 48 hours in both normal keratinocytes and oral cancer cells suggested that the phosphorylation of the two residues in Akt might result in a different biological response. Akt phosphorylation at S473 is induced by mTORC2 (mammalian target of rapamycin complex 2), but the association between TGFβ1 and mTORC2 is unknown [23]. By analysing our results along with the data obtained from another study we hypothesise that TGFβ1 can activate the PI3K-PDK1 complex that phosphorylates Akt at T308, increasing Akt kinase activity. Akt phosphorylates SIN1 at T86, enhancing mTORC2 kinase activity, which leads to phosphorylation of Akt S473 by mTORC2, thereby catalysing full activation of Akt [24].
In this study, wound healing (scratch) and scatter assays were included to investigate two different modes of cell migration. The wound healing (or scratch) assay is a popular 2-D in vitro assay to visualise collective cell migration and the scatter assay is used to visualise single cell migration. A modified gap closure assay was also employed in this study to avoid making wounds in the cell monolayer and to escape some confounding factors such as changes in the strength of cell–cell adhesions under different conditions and cell crowding that might affect cell migration [25]. Normal keratinocytes and oral cancer cells migrated out of cell colonies in response to addition of TGFβ1 as a single cell and with mesenchymal-type morphology. A mesenchymal-type cell shape with high vimentin and low/no E-cadherin expression further confirmed that TGFβ1-induced cell migration was accomplished by EMT. However, addition of Akt and MAPK phosphorylation inhibitors revealed a rather interesting result in the migration assays. Our data indicated that TGFβ1-induced oral cancer cell and normal keratinocyte migration through EMT is Akt and MAPK dependent, respectively. Previous studies showed that TGFβ1 stimulated both normal keratinocytes and oral cancer cell migration through EMT [26-29]. Secker et al (2008) also revealed that TGFβ1 stimulation of HaCaT cell migration through EMT is MAPK dependent [30]. A previous study also suggested that TGFβ-induced EMT in breast cancer cell was dependent on the PI3K/Akt signalling pathway [31]. This is the first report to show that TGFβ1-induced oral cancer cell migration is dependent on the Akt signalling pathway. This study also suggested that Akt phosphorylation played an important role in oral cancer cell migration, by the fact that it remained phosphorylated for 48 hours and this corresponded with the time for cell migration. On the other hand, the Akt inhibitor effectively blocked TGFβ1-induced oral cancer cell migration. Previous studies reported that Akt activates Twist, a basic helix-loop-helix transcription factor by phosphorylating the Ser16 residue, which induces EMT in HNSCC [32-34]. Although, in this study SMAD2/3 and MAPK were also phosphorylated in TGFβ1-induced oral cancer cells results indicated that these signalling molecules were not involved in cell migration. For example, SMAD2/3 phosphorylation persisted for only 2 hours, whereas oral cancer cell migration was observed after 24 hours and the MAPK inhibitor could not block this migration. This study therefore supports the idea that TGFβ1 could induce cell migration during both wound healing and cancer metastasis. The mode of cell migration and associated signalling cascade, however, largely depends upon the cell types, assay format, incubation time and available concentrations of the ligand.

Figure 7: An extrapolated diagram of the role TGFβ1-induced signalling pathways in oral cancer metastasis. Oral cancer cell migration is blocked by Akt inhibitor, MK-2206 which indicates oral cancer metastasis is Akt signalling pathway dependent. Normal keratinocytes migration, however, is dependent on MAPK signalling pathways. SMAD is phosphorylated in oral cancer cells but not involved in cell migration.
Conclusion
This study revealed that TGFβ1-induced normal keratinocyte and oral cancer cell migration is dependent on two separate non-SMAD dependent pathways, namely the MAPK and Akt signalling pathways, respectively. The result is summarised in an over simplified view of events in Figure 7. This study also suggests that two signalling pathways are involved in TGFβ1-induced wound healing (MAPK) and metastasis (Akt). Therefore, targeting the Akt signalling pathway to inhibit oral cancer metastases would be a potential approach in the clinic in future.
Acknowledgments
This work was conducted as part of a laboratory project for the degree of MRes in Oral Cancer and MSc in Oral Biology at the University of Dundee. The authors wish to thank Jacqueline Cox for her dedication and technical ability.
Contributions
Study concepts and design- MI, IE, SJ; Data acquisition- AA, BS, PS; Quality control of data, data analysis and interpretation- MI, AA, PS, BS; Manuscript preparation- MI; Manuscript editing and review- MI, SJ, IE.
Funding
This research did not receive any specific grant from funding agencies in the public, commercial, or not-for-profit sectors.
Ethical Approval
This article does not require any human/animal subjects to acquire any ethical approval.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Article Info
Article Type
Research ArticlePublication history
Received: Thu 11, Oct 2018Accepted: Tue 30, Oct 2018
Published: Mon 26, Nov 2018
Copyright
© 2023 Mohammad Islam. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited. Hosting by Science Repository.DOI: 10.31487/j.DOBCR.2018.03.004
Author Info
Corresponding Author
Mohammad IslamUnit of Cell and Molecular Biology, School of Dentistry, University of Dundee, Dundee, DD1 4HR, Scotland, UK
Figures & Tables
References
1. Johnson NW, Warnakulasuriya S, Gupta PC, Dimba E, Chindia M, et al. (2011) Global oral health inequalities in incidence and outcomes for oral cancer: causes and solutions. Adv Dent Res 23: 237-246. [Crossref]
2. Jou A, Hess J (2017) Epidemiology and Molecular Biology of Head and Neck Cancer. Oncol Res Treat 40: 328-332. [Crossref]
3. Leemans CR, Braakhuis BJ, Brakenhoff RH (2011 ) The molecular biology of head and neck cancer. Nat Rev Cancer 11: 9-22. [Crossref]
4. Vicente-Manzanares M, Webb DJ, Horwitz AR (2005) Cell migration at a glance. J Cell Sci 118: 4917-4919. [Crossref]
5. Rørth P (2009) Collective Cell Migration. Annu Rev Cell Dev Biol 25: 407-429. [Crossref]
6. Thiery JP (2002) Epithelial–mesenchymal transitions in tumour progression. Nat Rev Cancer 2: 442-454. [Crossref]
7. Schor SL (1994) Cytokine control of cell motility: modulation and mediation by the extracellular matrix. Prog Growth Factor Res 5: 223-548. [Crossref]
8. Ellis IR, Schor AM, Schor SL (2007) EGF AND TGF-alpha motogenic activities are mediated by the EGF receptor via distinct matrix-dependent mechanisms. Exp Cell Res 313: 732-741. [Crossref]
9. Islam MR, Jones SJ, Macluskey M, Ellis IR (2014) Is there a pAkt between VEGF and oral cancer cell migration? Cell Signal 26: 1294-1302. [Crossref]
10. Islam, Mane S, Hyder E, Jones S, Ellis I (2017) The motogenic effect of EGF and TGF-α on the migration of tumor cells from the oral region. Transl Res Oral Oncol 2: 2057.
11. Weeks BH, He W, Olson KL, Wang XJ (2001) Inducible expression of transforming growth factor beta1 in papillomas causes rapid metastasis. Cancer Res 61: 7435-7443. [Crossref]
12. Fukai Y, Fukuchi M, Masuda N, Osawa H, Kato H, et al. (2003) Reduced expression of transforming growth factor-beta receptors is an unfavorable prognostic factor in human esophageal squamous cell carcinoma. Int J Cancer 104: 161-166. [Crossref]
13. Pasini FS, Brentani MM, Kowalski LP, Federico MH (2001) Transforming growth factor beta1, urokinase-type plasminogen activator and plasminogen activator inhibitor-1 mRNA expression in head and neck squamous carcinoma and normal adjacent mucosa. Head Neck 23: 725-732. [Crossref]
14. Lu S-L, Reh D, Li AG, Woods J, Corless CL, et al. (2004) Overexpression of transforming growth factor beta1 in head and neck epithelia results in inflammation, angiogenesis, and epithelial hyperproliferation. Cancer Res 64: 4405-4410. [Crossref]
15. Derynck R, Zhang YE (2003) Smad-dependent and Smad-independent pathways in TGF-β family signalling. Nature 425: 577-584. [Crossref]
16. Moustakas A, Heldin CH (2005) Non-Smad TGF-beta signals. J Cell Sci 118: 3573-3584. [Crossref]
17. Moustakas A, Heldin CH (2007) Signaling networks guiding epithelial-mesenchymal transitions during embryogenesis and cancer progression. Cancer Sci 98: 1512-1520. [Crossref]
18. Xu J, Lamouille S, Derynck R (2009) TGF-β-induced epithelial to mesenchymal transition. Cell Res 19: 156-172. [Crossref]
19. Katsuno Y, Lamouille S, Derynck R (2013) TGF-β signaling and epithelial–mesenchymal transition in cancer progression. Curr Opin Oncol 25: 76-84. [Crossref]
20. Chen HC (2005) Cell-scatter assay. Methods Mol Biol 294: 69-77. [Crossref]
21. Rodriguez LG, Wu X, Guan JL (2005) Wound-healing assay. Methods Mol Biol 294: 23-29. [Crossref]
22. Zi Z, Chapnick DA, Liu X (2012) Dynamics of TGF-β/Smad signaling. FEBS Lett 586: 1921-1928. [Crossref]
23. Sarbassov DD, Guertin DA, Ali SM, Sabatini DM (2005) Phosphorylation and Regulation of Akt/PKB by the Rictor-mTOR Complex. Science 307: 1098-1101. [Crossref]
24. Yang G, Murashige DS, Humphrey SJ, James DE (2015) A Positive Feedback Loop between Akt and mTORC2 via SIN1 Phosphorylation. Cell Rep 12: 937-943. [Crossref]
25. Cory G (2011) Scratch-Wound Assay. In: Claire W, Maddy P, editors. Cell Migr. Dev. Methods Protoc. Totowa, NJ: Humana Press 25-30.
26. Räsänen K, Vaheri A (2010) TGF-beta1 causes epithelial-mesenchymal transition in HaCaT derivatives, but induces expression of COX-2 and migration only in benign, not in malignant keratinocytes. J Dermatol Sci 58: 97-104. [Crossref]
27. Hatta M, Miyake Y, Uchida K, Yamazaki J (2018) Keratin 13 gene is epigenetically suppressed during transforming growth factor-β1-induced epithelial-mesenchymal transition in a human keratinocyte cell line. Biochem Biophys Res Commun 496: 381-386. [Crossref]
28. Takayama Y, Mori T, Nomura T, Shibahara T, Sakamoto M (2010) Parathyroid-related protein plays a critical role in bone invasion by oral squamous cell carcinoma. Int J Oncol 36: 1387-1394. [Crossref]
29. Quan J, Elhousiny M, Johnson NW, Gao J (2013) Transforming growth factor-β1 treatment of oral cancer induces epithelial-mesenchymal transition and promotes bone invasion via enhanced activity of osteoclasts. Clin Exp Metastasis 30: 659-670. [Crossref]
30. Secker GA, Shortt AJ, Sampson E, Schwarz QP, Schultz GS, et al. (2008) TGFbeta stimulated re-epithelialisation is regulated by CTGF and Ras/MEK/ERK signalling. Exp Cell Res 314: 131-142. [Crossref]
31. Chen L, Fu H, Luo Y, Chen L, Cheng R, et al. (2017) cPLA2α mediates TGF-β-induced epithelial–mesenchymal transition in breast cancer through PI3k/Akt signaling. Cell Death Dis 8: 2728-2728. [Crossref]
32. Way T-D, Huang J-T, Chou C-H, Huang C-H, Yang M-H, et al.( 2014) Emodin represses TWIST1-induced epithelial–mesenchymal transitions in head and neck squamous cell carcinoma cells by inhibiting the β-catenin and Akt pathways. Eur J Cancer 50: 366-378. [Crossref]
33. Tang H, Massi D, Hemmings BA, Mandalà M, Hu Z, et al. (2016) AKT-ions with a TWIST between EMT and MET. Oncotarget 7:62767-62777. [Crossref]
34. Jiang L, Wang Z, Liu C, Gong Z, Yang Y, et al. (2017) TrkB promotes laryngeal cancer metastasis via activation PI3K/AKT pathway. Oncotarget 8: 108726-108737. [Crossref]