Helicobacter pylori Elicits B7-H3 Expression on Gastric Epithelial Cells: Implications in Local T Cell Regulation and Subset Development During Infection
A B S T R A C T
Helicobacter pylori (H. pylori) is a gram-negative bacterium that infects more than 50% of humanity and is associated with gastritis, peptic ulcer and gastric cancer. Although CD4+ T cells are recruited to the gastric mucosa, the host is unable to clear the bacteria. Previously, we demonstrated that H. pylori infection upregulates the expression of the T cell co-inhibitory molecule B7-H1 while simultaneously downregulating the expression of T cell co-stimulatory molecule B7-H2 on gastric epithelial cells (GEC), which together affect the Treg and Th17 cell balance and foster bacterial persistence. Because B7-H3, another member of the B7 family of co-inhibitory receptors, has been found to have important immunoregulatory roles and in cancer, in this study we examined the expression of B7-H3 molecules on GEC and how the expression is regulated by H. pylori during infection. Our study showed that both human and murine GEC constitutively express B7-H3 molecules, but their expression levels increased during H. pylori infection. We further demonstrated that H. pylori uses its type 4 secretion system (T4SS) components CagA and cell wall peptidoglycan (PG) fragment to upregulate B7-H3. Th17 cells and Treg cells which are increased during H. pylori infection also had an effect on B7-H3 induction. The underlying cell signaling pathway involves modulation of p38MAPK pathway. Since B7-H3 were shown to up-regulate Th2 responses, the phenotype of T cell subpopulations in mice infected with H. pylori PMSS1 (contains functional T4SS) or SS1 (cannot deliver CagA into GEC) strains were characterized. A mixed Th1/Th2 response in H. pylori infected mice was observed. Consistent with previous findings, increased Treg cells and decreased Th17 cells in MLN of PMSS1 infected mice compared to SS1 infected mice was observed. Human biopsy samples collected from gastritis biopsies and gastric tumors showed a strong association between increased B7-H3 and Th2 responses in H. pylori strains associated with gastritis. T cell: GEC co-cultures and anti-B7-H3 blocking Ab confirmed that the induction of Th2 is mediated by B7-H3 and associated exclusively with an H. pylori gastritis strain not cancer or ulcer strains. In conclusion, these studies revealed a novel regulatory mechanism employed by H. pylori to influence the type of T cell response that develops within the infected gastric mucosa.
Keywords
Helicobacter pylori (H. pylori), B7-H3, gastric epithelial cells (GEC) , Th2
Introduction
Helicobacter pylori (H. pylori) colonizes the human gastric mucosa and may induce gastritis, peptic ulcer and two forms of neoplasia: gastric adenocarcinoma and mucosa-associated lymphoid tissue (MALT) lymphoma [1]. Epidemiological data suggest that 60-90% of gastric cancer cases are caused by H. pylori [2, 3]. Patients infected with CagA (cytotoxin associated gene A)-positive H. pylori strains have an elevated risk of developing peptic ulcer and gastric cancer [4, 5]. CagA is the only known effector protein produced by the H. pylori cag PAI (cag pathogenicity island), which is a 40 KDa chromosomal region that contains the genes that code for structural components of the type 4 secretion system (T4SS). T4SS is a molecular syringe-like structure. Upon attachment of H. pylori to gastric epithelial cells (GEC), CagA is injected via the T4SS and consequently becomes phosphorylated in the tyrosine residue of their EPIYA motifs by host Src kinases and c-Ab1 [6-10]. Both phosphorylated and unphosphorylated forms of CagA can interact with a range of host cell signaling proteins and activates them, which results in several physiological changes in GECs [11-13]. CagA alone has been shown to act as a oncoprotein since transgenic mice expressing H. pylori CagA develop multiple types of neoplasms [62]. In addition to CagA, H. pylori also translocates via the T4SS its cell wall peptidoglycan (PG) fragments, which are recognized by intracellular pattern recognition receptor NOD1 and activates MAPKs and NFkB pathways [14-16].
B7-H3 (CD276) is a newer member of the B7 family that shares 20–27% identical amino acids with other members of this family of receptors [17]. Human B7-H3 protein is not constitutively expressed but can be induced in activated dendritic cells, B cells, T cells, NK cells and in some tumor cell lines [17-20]. B7-H3 has been shown to be strongly expressed in unstimulated tracheal, bronchial, and alveolar epithelial cells, and the expression was induced by respiratory syncytial virus (RSV) infection [21]. B7-H3 was initially identified as a co-stimulatory molecule that was shown to promote T-cell proliferation and IFN-γ production [17]. However, recent studies have presented contradictory roles for B7-H3, since they suggest that B7-H3 has both immunological stimulatory and inhibitory functions [17-20, 22-25]. For instance, in conjunction with anti-CD3, B7-H3-Ig fusion protein co-stimulates CD4+ and CD8+ T cells and induces IFN-γ production. Other independent studies demonstrated that acute and chronic cardiac allograft rejection is reduced in B7-H3 knockout mice, which further support a stimulatory role for B7-H3 on T cells [25]. In contrast, B7-H3 has been reported to impair T-helper (Th)1 cell responses and inhibit cytokine production [22]. An in vivo study also showed an inhibitory role of B7-H3 [19, 22, 24]. B7-H3 not only affects T cell activation /inactivation but a recent study in an asthma model showed that B7-H3 also plays a role in the induction of Th2 cells [26]. Moreover, other than its role in regulating T cell activity and subset development, it may also serve as a biomarker for tumor progression and development of cancer. Higher expression of B7-H3 has been shown in different types of cancer [27-31]. An increased expression of B7-H3 was reported to lead to an increased risk of recurrence of some cancers, while increased B7-H3 expression is in sometimes linked with prospective survival in other cancers [27-31]. Recently increased B7-H3 expression was shown in circulating tumor cells in gastric cancer patients compared to healthy volunteers. Moreover, patients with increased B7-H3 levels showed lower survival rates [32]. However, a separate study reported that increased B7-H3 during gastric cancer was associated with increased survival rate [31]. Together, these observations suggested that B7-H3 might be also involved in cancer immunity and B7-H3 may also influence cancer progression beyond its immunoregulatory roles.
H. pylori usually causes chronic infection. Though the host mounts an increased CD4+ T cell response but those T cells are hyporesponsive. During H. pylori infection, patients have a mixed Th1/Th2 response, with increased Treg and Th17 cells in their circulation [33-39]. Though there are reports showing the type of T cell responses elicited by H. pylori infection, there is a gap in our knowledge regarding the mechanism that H. pylori uses to induce different phenotypic subsets of T cells. Previously our group has shown that H. pylori modulates B7 molecule expression in GECs, which not only help restrain T cells responses, but also induce T regulatory (Treg) cells to assist in H. pylori survival [40-42]. Our data also showed that H. pylori uses its T4SS to downregulate B7-H2 expression in GEC, which helps to keep Th17 cells in suboptimal levels, since Th17 are important in the control of extracellular bacterial infections, this downregulation of B7-H2 helps H. pylori to persist [41]. We also demonstrated that H. pylori-mediated up-regulation of B7-H1 expression in GEC causes induction of Treg cells, which contributes to the establishment of a chronic infection [42]. In this study, we investigated another important B7 molecule, B7-H3 and showed that H. pylori upregulates the expression of this molecule on GEC. The upregulation of B7-H3 is regulated not only by the T4SS but also by the cytokines produced by Th17 and Treg cells. We further evaluated the underlying cell signaling pathway and demonstrate that H. pylori uses the p38 MAPK pathway for B7-H3 upregulation.
H. pylori is one of the most genetically diverse bacterial species. H. pylori strains differ in the rate with which they have cag PAI in their genome. The EPIYA motifs in cagA gene also differs between Asian and western countries. Moreover, H. pylori infection may result in gastritis, ulcer and gastric cancer development. We examined how H. pylori strains isolated from these three types of gastric diseases modulate B7-H3 expression on epithelial cells. In this study we were interested to determine whether the increase of B7-H3 is consistent with all strains or not. Using different H. pylori strains and patient samples from gastritis and tumors we have shown that only H. pylori strains associated with gastritis causes increased B7-H3 expression and induction of the GATA3+ Th2 cell response. This finding was further confirmed by co-culturing GECs infected with different H. pylori strains with naïve CD4+ T cells. This is a novel finding which shows how H. pylori manipulates GECs to differentially express the B7-H3 molecules and thus regulates T cell responses involved in the H. pylori associated immune-pathogenesis to promote bacterial persistence. Future studies will examine how these findings may be applied in vaccine efforts against H. pylori and possibly in prevention or treatment of gastric cancer.
Material and Methods
I. Human tissue
Gastric antrum biopsy specimens were obtained from consenting patients undergoing gastro-esophageal-duodenoscopy in accordance with an approved Institutional Review Board protocol. GECs were isolated from the biopsy specimens as described previously [41]. Patients were considered infected if H. pylori was detected by rapid urease testing, histopathology, and by culture of H. pylori from biopsies.
II Cell lines, bacterial cultures and small peptides
Human GECs N87 and AGS were obtained from the American Type Culture Collection (ATCC) and HGC-27 was obtained from RIKEN, The Institute of Physical and Chemical Research, Japan. All cell lines were maintained in RPMI 1640 with 10% fetal bovine serum (FBS) and 2 mM L-glutamine. As representative murine GEC, Immortomouse stomach epithelium (ImSt) cells were maintained in media described by Whitehead et al. [43]. H. pylori strains 51B and 26695 as well as their corresponding isogenic cagA and cag PAI mutants were described previously [41, 44]. H. pylori LC-11 and CA8 were originally isolated from the antral mucosa of a patient with duodenal ulcer and gastric cancer, respectively, as previously described, Tryptic soy agar (TSA) plates supplemented with 5% sheep’s blood (Becton Dickinson, San Jose, CA) were used to grow H. pylori strains [45, 46]. Blood agar plates with 2.5 μg/ml of chloramphenicol (Technova, Hollister, CA) were used to maintain cagA- and cag PAI- strains at 37°C under microaerophilic conditions [41]. For the infection of mice H. pylori Sydney strain 1 (SS1) and PM-SS1 (pre-mouse SS1) were used, which were provided by Drs. J. Pappo (Astra) and Richard Peek (Vanderbilt Univ.), respectively [47]. iEDAP (InvivoGen, San Diego, USA), a PG-like molecule that is a NOD1 ligand, was used to investigate the role of PG in B7-H3 expression.
III Animals
Female six-to-eight week old C57BL/6 mice (Jackson Laboratory, Bar Harbor, ME) were used in the model of gastric H. pylori infection. Animals were tested negative for the intestinal Helicobacter spp. prior to their use in the experiments.
IV. Flow Cytometry
APC-conjugated anti-human B7-H3 (clone 185504) and isotype controls were purchased from R&D Systems. T cells from co-culture assays, described below, were stained for CD25, FoxP3, RORγ, Tbet and GATA3 for analysis by flow cytometry using a protocol described previously [42]. Mouse anti-human CD25-PECy7, FoxP3-Alexafluor 488, Tbet-PerCPCy5.5, Gata3-eFluor 660 were used for staining. The viability dye eFluor 780 (eBioscience, San Diego, CA, USA) was included in the experiments to gate on viable cells. Cells were analyzed by flow cytometry on a LSRII instrument. The data were analyzed with BD FACSDiva software (BD Biosciences, San Jose, CA) and FlowJo (Tree Star, Inc, Ashland, OR).
V Cell signaling inhibitors
NFκB inhibitor, CAY10512 (10 µM; Cayman Chemical, MI); JAK/STAT3 inhibitor AG-490 (100 ng/mL; Enzo Life Sciences, Farmingdale, NY), PI3K inhibitor, Wortmannin (100 nM; Calbiochem, Billerica, MA); and p38 MAPK inhibitor, PD169316 (10 μM/mL; Cayman Chemical, MI) were used to inhibit intracellular signaling.
VI Real-time RT-PCR
Real-time RT-PCR analysis was performed as previously described [41].
VII Murine infection and detection of B7-H3, FoxP3, RORyt, Tbet and GATA3 expression
C57BL/6 mice were orogastrically inoculated with 108 CFU (in 100 µL of PBS/inoculation) of H. pylori SS1 or PMSS1 strains, three times over a week. Four weeks later mice were euthanized, mesenteric lymph node (MLN) were removed, homogenized, mRNA was isolated and expression of FoxP3, RORγt, Tbet, GATA3, IL-10, IFN-γ, IL-4 and IL-17A were determined using RT-PCR.
VIII T cell isolation and co-culture with GEC
Naïve CD4+ T cells were isolated from human peripheral blood as previously described [48]. GEC-T cell co-cultures were established as described earlier [41]. Briefly, GECs were preinfected with H. pylori CA8 (cancer strain), H. pylori 51B (gastritis strain) and H. pylori LC-11 (ulcer strain). After 8 h of infection GECs were washed and co-cultured with 1x106 T cells to obtain 3:1 T cell:GEC ratio and incubated for 5 days at 37º C with 5% CO2. For blocking, anti-B7-H3 blocking antibody or isotype (rat IgG2a κ) control (1 μg/mL, functional grade from eBioscience) were added to GECs 1 h before co-culture.
IX. Bio-Plex
The levels of IL-4 from T cell-GEC co-culture were measured using Luminex array (Millipore, Billerica, MA, USA) according to the manufacturer´s instruction. Samples were analyzed using Bio-Plex Manager software (Bio-Rad).
X. Statistical analysis
The results were expressed as the mean ± SE of data obtained from at least three independent experiments done with triplicate sets per experiment unless otherwise indicated. Differences between means were evaluated by analysis of variance (ANOVA) using student t test for multiple comparisons and considered significant if p was <0.05.
Results
I Expression of B7-H3 on gastric biopsies
To determine the expression of B7-H3 in relation to H. pylori infection we isolated GECs from biopsy samples, which were collected from H. pylori infected or from healthy individuals. B7-H3 expression was measured by real time RT-PCR after mRNA was extracted from the samples. Our RT-PCR data showed a strong upregulation of B7-H3 expression in the H. pylori infected biopsies compared to uninfected samples (Figure 1A).
II H. pylori T4SS regulate B7-H3 expression on GEC during infection
To evaluate whether B7-H3 upregulation is a direct effect of H. pylori infection and not an indirect result of inflammatory changes in the host, we used GEC lines and infected them with H. pylori. Since H. pylori T4SS has the capacity to modulate GEC homeostasis and because we have seen their effect in the modulation of B7-H1 and B7-H2 molecules, we used H. pylori 51B wild type (WT) and H. pylori 51B cag PAI mutant strain to infect GEC (N87 cells) [41, 49]. B7-H3 expression was measured after 24-hr infection using flow cytometry. A significant upregulation of B7-H3 expression in GEC infected with H. pylori WT but not with H. pylori cag PAI mutant strains was observed, suggesting that H. pylori T4SS plays role in B7-H3 induction (Figure 1B). To further dissect the role of the effector protein CagA H. pylori 51B cagA mutant was used to infect GEC along with the H. pylori 51B WT strain. Both flow cytometry and RT-PCR data showed that CagA influences B7-H3 upregulation, since in the absence of CagA B7-H3 expression by GECs remained at basal levels (Figure 1C, D). These results were also confirmed in different cell lines (AGS, HGC-27) and by using H. pylori 26695 WT and the corresponding isogenic mutant strains (not shown). Furthermore, a murine cell line was used to confirm these findings and to evaluate whether murine GEC express B7-H3 and whether this expression is regulated by H. pylori T4SS or not, before using a murine model. To this end, the murine GEC line (ImSt) were infected with H. pylori PMSS1, which contains a functional T4SS and with H. pylori SS1 strain in which the T4SS is defective and cannot deliver CagA into GEC. Flow cytometry data showed a significant upregulation of B7-H3 expression in murine GEC infected with H. pylori PMSS1 strain but not with the SS1 strain (Figure 1E). Overall, these data demonstrated a strong correlation between the presence of T4SS, more specifically of the CagA oncoprotein, and induction of B7-H3 expression on GEC.

Figure 1: H. pylori T4SS up-regulates B7-H3 expression on GECs. (A) Gastric biopsy samples were collected from H. pylori-positive patients and healthy individuals, GECs were collected and analyzed for B7-H3 mRNA expression by real-time RT-PCR. N87 cells were infected with (B) H. pylori 51B WT and cag PAI- or with (C) H. pylori 51B WT and cagA- for 24 h. The surface expression of B7-H3 was determined by using immunostaining followed by flow cytometry. (D) N87 cells were infected with H. pylori 51B WT and cagA- for 2 h, and B7-H3 mRNA expression was analyzed by using RT-PCR. mRNA levels for B7-H3 were normalized to 18S and compared to the level of B7-H3 mRNA of untreated N87 cells. (E) Murine GECs (ImSt) were infected for 24 h with H. pylori PMSS1, which has a functional CagA delivery system, or with H. pylori SS1, lacking a CagA delivery system. Surface expression of B7-H3 was determined by flow cytometry. The data were expressed as a percentage of positive cells. The means ± SD are shown as the results of duplication of one of four representative experiments, n=8, *P < 0.05.
III Role of PG in B7-H3 upregulation
Along with CagA, H. pylori T4SS also translocates PG fragments into GECs, which are recognized by NOD1 and cause activation of cell signaling pathways that result in inflammatory mediator release [14-16, 50]. Further, in H. pylori infection, NOD1 is up-regulated and associated with higher inflammation in GC [63]. To determine the involvement of PG in B7-H3 upregulation GECs were treated with iEDAP, which is a PG analogue recognized by NOD1 ligand. B7-H3 expression was significantly upregulated in mRNA level (Figure 2A) after iEDAP stimulation. Flow cytometry was used as an independent approach to measure the upregulated surface expression of B7-H3 (Figure 2B). Kinetics data showed a progressive upregulation of B7-H3 as early as 18-hr of stimulation which peaked at 24-hr (Figure 2C) and is mediated by H. pylori T4SS component CagA and PG (Figure 2D).
IV H. pylori uses p38MAPK pathway for B7-H3 upregulation
Further analysis was done to determine the cell signaling pathway used by H. pylori for B7-H3 up-regulation. To that end, the cells were treated with different pharmacological inhibitors of NFκB, MAPK, STAT3, PI3K and mTOR pathways. Our data indicated that upregulation of B7-H3 by the H. pylori strain was blocked in the presence of PD169316, which is a p38 MAPK specific inhibitor (Figure 3). In contrast, inhibition of PI3K, mTOR, STAT3 and NFκB pathways did not affect H. pylori mediated upregulation of B7-H3 expression (Data not shown). These results suggest that p38 MAPK pathway is a key signaling pathway in H. pylori-mediated upregulation of B7-H3 on GECs.
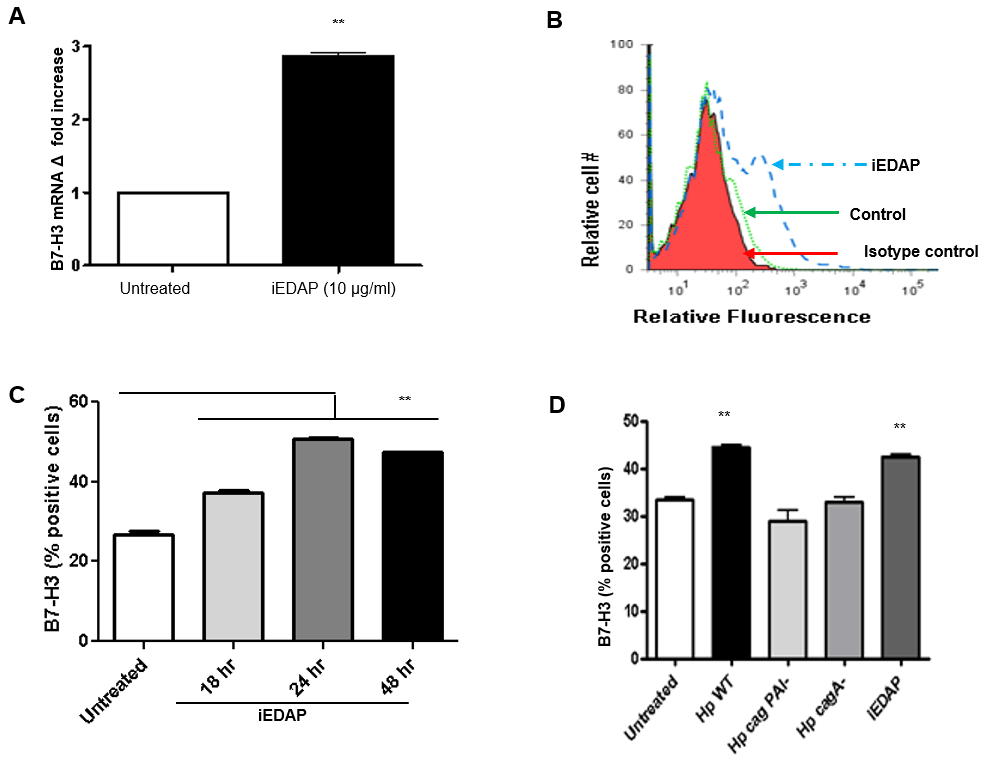
Figure 2: H. pylori T4SS translocated PG causes induction of B7-H3 expression by GECs. (A) B7-H3 mRNA expression was analyzed by using real-time quantitative RT-PCR in N87 cells. RNA was isolated from untreated and 2 h iEDAP (dipeptide present in peptidoglycan) treated (10 µg/mL) cells. mRNA levels for B7-H3 were normalized to 18S and compared to the levels of B7-H3 mRNA in untreated N87 cells. N=9, *P < 0.05. (B) Flow cytometric analysis of GEC (N87) cells stained for B7-H3 after exposure to 10 µg/mL iEDAP for 24 h (in a representative histogram for AGS cells where the solid peak is the isotype control) or (C) for different times (18, 24 and 48 h) showed increased expression. (D) N87 cells were infected with H. pylori WT, H. pylori cag PAI, and H. pylori cagA- and stimulated with iEDAP for 24 h and B7-H3 expression was measured by flow cytometry. The means are shown as the results of duplicates in four experiments, n= 8,*P < 0.05.
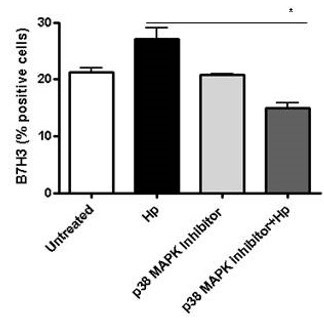
Figure 3: B7-H3 up-regulation by H. pylori depends on p38 MAPK pathway. B7-H3 expression on GEC was measured by flow cytometry after treating the cells with p38 MAPK inhibitor (PD169316 10 μM/ml) for 1 h and infected with H. pylori for 24 h. The means ± SD are shown as the results of duplicates in four experiments, n=8, * P < 0.05, ** P < 0.01 and *** P < 0.001.
V. B7-H3 expression is regulated by Th17 and Treg cells
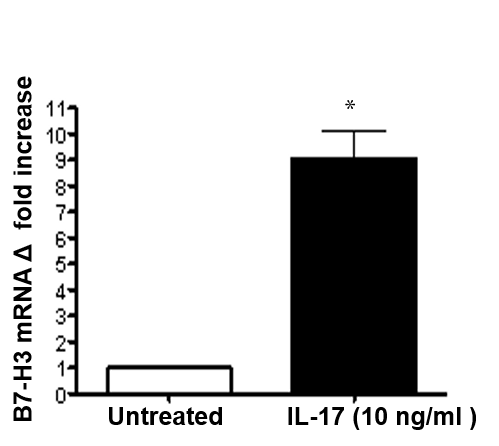
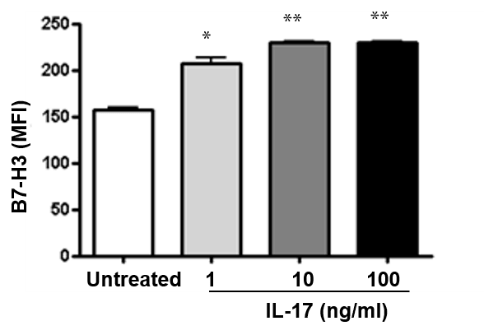
Cytokines regulate the expression of immunoregulatory molecules, which allows for fine tuning of the immune response. During H. pylori infection there is induction of Th17 cells [37-39]. Since patients have increased circulating levels of IL-17 we sought to investigate the effect of this cytokine on B7-H3 expression. In RT-PCR analysis (Figure 4A) significant induction of B7-H3 after IL-17 (10 ng/ml) stimulation was observed. Further, the experiments showed that the expression of B7-H3 on the GECs in response to IL-17 stimulation was increased in a dose-dependent manner (1-100 ng/ml) (Figure 4B). The surface expression of this ligand was also analyzed at different time points (18-hr, 24-hr and 48-hr) after IL-17 treatment. Expression was significantly increased in GECs after 18-hr of incubation with IL-17, which remains constant after 24 h but decreases after 48 h incubation (Figure 4C).
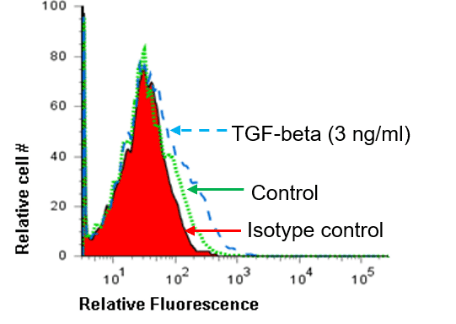
Treg cells, which are frequently found in H. pylori-infected patients, produce IL-10 and TGF-β [34, 35]. Since there is bidirectional regulation of Treg cells and B7-H1, we investigated whether the hallmark cytokines produced by these cells affect B7-H3 expression [51]. To that end we stimulated GEC with either IL-10 or TGF-β alone or in combination. Both IL-10 and TGF-β induced B7-H3 expression on GECs (Figure 5A, B). Flow cytometry data also showed a cumulative effect of IL-10 and TGF-β in B7-H3 expression (Figure 5C). Taken together, these data suggested that cytokines produced by Th17 and Treg cells play an important role in B7-H3 expression in GEC. Thus H. pylori regulates B7-H3 expression both directly by using CagA cytotoxin and also indirectly by inducing these T cell subtypes.
Figure 4: B7-H3 expression is regulated by IL-17. (A) GEC (N87) cells were treated with IL-17 (10 ng/ml) for 2 h and B7-H3 expression was measured by RT-PCR. mRNA levels for B7-H3 was normalized to 18S and compared to the levels of B7-H3 mRNA of untreated N87 cells. Kinetics and dose response of IL-17-mediated B7-H3 up-regulation was determined by treating GEC (N87) cells with (B) different concentrations (1, 10 and 100 ng/ml) of IL-17 for 24 h or (C) exposing the GEC (N87) cells to IL-17 (10 ng/ml) for different time points (18, 24 and 48 h) and measuring the B7-H3 expression by flow cytometry. The data were expressed as mean fluorescence intensity (MFI). The means ± SD are shown as the result of duplicates of one of four representative experiments: n=8, * P < 0.05, ** P < 0.01 and *** P < 0.001.
Figure 5: B7-H3 expression is regulated by Treg cell cytokines. (A) Flow cytometry analysis of GEC cells stained for B7-H3 after exposure to 5 ng/mL IL-10 for 24 h showed increased expression in a representative histogram where the solid peak is the isotype control (B) Flow cytometry was done to measure B7-H3 expression on GECs after treating the cells with TGF-β (3 ng/ml) for 24 h. (C) Flow cytometry was done to measure B7-H3 expression on GECs treated with either IL-10 (5 ng/mL) or TGF-β (3 ng/ml) or both IL-10 and TGF-β. The data were expressed as the percentage of positive cells. The means ± SD are shown as the results of duplicates of one of four representative experiments: n=8, * P < 0.05, ** P < 0.01 and *** P < 0.001.
VI. Different T cell subset development during H. pylori infection
During H. pylori infection there is an increased frequency of Th1/Th17 and Treg cells in the gastric mucosa. Previously we have shown that H. pylori infection upregulates B7-H1 molecule expression by GECs, which in turn helps to induce further Treg cell development [49]. On the other hand, we also showed that H. pylori T4SS mediated downregulation of B7-H2 in GEC, which impairs Th17 cell development [41]. Besides the reported effects of B7-H3 on T cell activation and inactivation, recent studies by Nagashima O et al., showed that B7-H3 can upregulate Th2 responses [26]. Since our data showed H. pylori upregulates B7-H3 expression we sought to investigate whether the modulation of B7-H3 expression affects locaßl T cell responses. To that end, we collected MLN from mice infected with PMSS1 and SS1 strains and analyzed the T cell subsets present by measuring mRNA expression of the different T cell transcription factors considered “master regulators” for each CD4+ T cell subset, such as GATA3, Tbet, RORγ and FoxP3 for Th2, Th1, Th17 and Treg cells, respectively. Mice infected with SS1 strain showed increased induction GATA3, Tbet and RORγ compared to the PMSS1 strain. However, the MLN cells from PMSS1 infected mice showed increased FoxP3 expression compared to those from SS1 infected mice (Figure 6A). The mRNA expression of the corresponding cytokines produced by Th2, Th1, Th17 and Treg cells, e.g. IL-4, IFN-γ, IL-17A and IL-10, in MLN was further measured. The cytokine data correlate with the transcription factors found in mice infected with the different strains (Figure 6B).

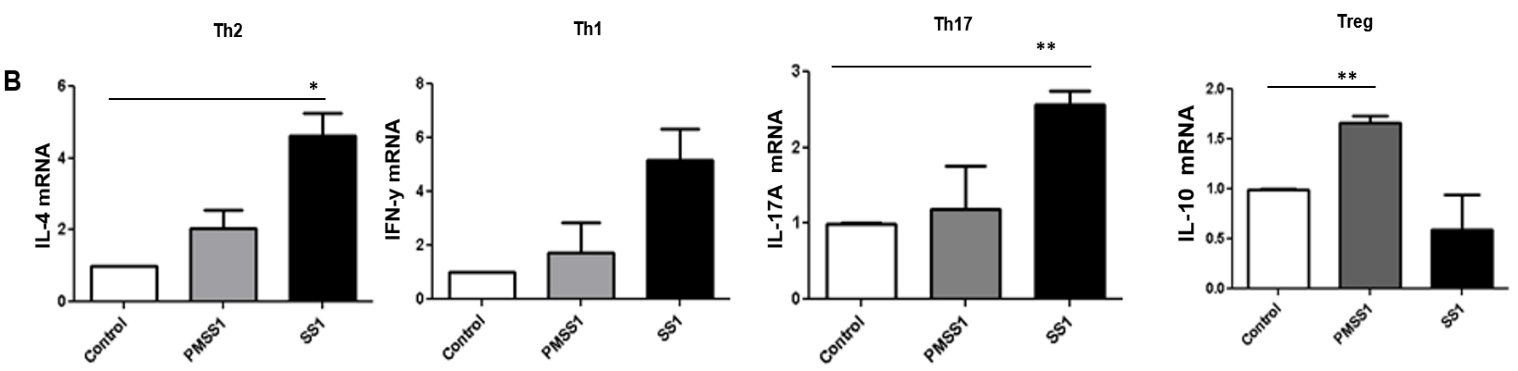
Figure 6: Different T cell subsets developed during H. pylori infection. C57BL/6 mice were challenged with H. pylori strain PMSS1 or with H. pylori SS1. Mice were sacrificed after 4 weeks of infection, MLN were collected, and expression measured of (A) GATA3, Tbet, RORγt, FoxP3 and (B) IL-4, IFN-γ, IL-17A, IL-10 mRNA by RT-PCR. Y-axis in each panel represents the fold increase in mRNA expression. Five mice per group were used in this experiment.
VII. Increased B7-H3 and GATA3 expression in gastritis patients
A previous report showed the presence of Th2 cells during H. pylori infection [31]. Consistent with that report, herein, the mouse model also showed induction of GATA3+ Th2 cell in MLN after H. pylori infection. Since B7-H3 has been shown to influence Th2 cell development, we sought to determine the influence of B7-H3 induction by GECs during H. pylori infection in Th2 cells response and whether it depends on the infecting strain. To this end, specimens from patients with gastritis and gastric tumors were evaluated. Biopsy samples from gastritis and samples from gastric tumors were evaluated for the relative expression of B7-H3 and GATA3. Interestingly, samples collected from gastritis patients showed increased B7-H3 and GATA3 expression compared to those from healthy individuals. However, in the case of patients with gastric tumors the expression of both B7-H3 and GATA3 was decreased, which suggested B7-H3 and Th2 induction during H. pylori infection might be a characteristic of gastritis strains (Figure 7).

Figure 7: B7-H3 and Th2 induction is associated with gastritis. B7-H3 and GATA3 expression in biopsy and tumor samples isolated from gastritis or gastric tumor patients with a history of H. pylori infection.
VIII. B7-H3 expressed by GEC after H. pylori infection induces development of Th2 cells
To further confirm whether the induction of B7-H3 and Th2 is only associated with H. pylori gastritis strains, N87 cell lines were treated with either medium alone or with different H. pylori strains: CA8 (from a gastric cancer case), 51B (from a gastritis case) and LC-11 (from an ulcer case) [44, 45, 61]. After 8 h of infection, the cells were washed extensively and incubated with isolated CD4+ naïve T cells for 5 days. T cells were harvested and stained for CD25, Tbet, GATA3, RORγt and FoxP3 monoclonal antibodies and analyzed by flow cytometry. The data showed increased GATA3+ cells in T cells co-cultured with GECs pre-infected with the gastritis strain (H. pylori 51B), but not with the other strains (Figure 8A). A significant increase in GATA3+ Tbet+ double-positive cells was also observed in T cells co-cultured with GECs pre-infected with the gastritis strain, suggesting conversion of Th1 cells to Th2 cell type (data not shown). Interestingly, incubation of the T cells with GECs pretreated with blocking B7-H3 antibody reduced Th2 cell frequency. This data suggested that induction of Th2 is influenced by B7-H3 (Figure 8B).
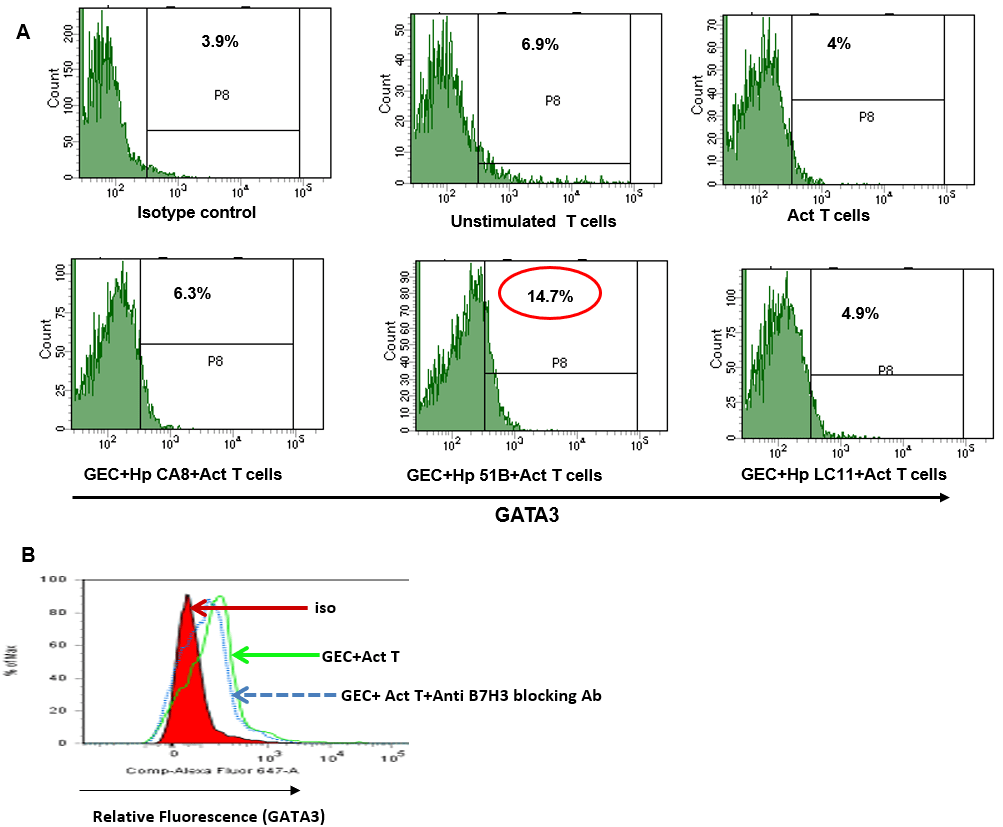
Figure 8: Increased B7-H3 expression and Th2 induction by H. pylori gastritis strain. (A) GECs were infected with either H. pylori CA8/51B/LC-11 strains for 8 h, washed and co-cultured with T cells. Cells were stained and GATA3 expression analyzed by flow cytometry. (B) A representative histogram showing GATA3 expression by GEC co-cultured with activated T cells in the presence or absence of ant-B7-H3 blocking Ab.
Discussion
B7-H3 has previously been considered a co-stimulatory molecule which promotes T cell proliferation [17]. But later studies have shown that B7-H3 can also function as a co-inhibitory molecule [17-20, 22-25]. B7-H3 is expressed by an array of cell types. A previous study showed that RSV causes induction of B7-H3 in tracheal, bronchial, and alveolar epithelial cells [21]. We have shown previously that B7-H3 is expressed by GEC [40]. In this study we demonstrate that H. pylori increases the expression of B7-H3 on GEC upon infection. Increased expression of B7-H3 was demonstrated on the GECs isolated from biopsies of H. pylori infected patients. This observation was confirmed in vitro by increased B7-H3 mRNA level and surface expression in a panel of GEC lines (N87, AGS and HGC-27) after infecting with H. pylori 51B and 26695 strains. Although our lab previously showed that GEC express B7-H3, the expression was unchanged after infection with H. pylori LC-11 strain [40]. That observation together with our recent observations suggest that this cellular response to infection might depend on the infecting H. pylori strain since both 51B and 26695 were isolated from patients with gastritis, while LC-11 originated from a patient with peptic ulcer. These observations were further confirmed in this study by using gastric tissue samples from patients with different gastric diseases associated with H. pylori infection.
H. pylori T4SS is an important virulence factor that influences GEC homeostasis [52, 53]. The recent findings by our group regarding the involvement of T4SS by H. pylori to modulate B7 molecule expression, led us to consider H. pylori T4SS as a virulence factor responsible for the up-regulation of B7-H3 by GECs [41]. By using H. pylori WT and cag PAI isogenic mutants, we showed here that B7-H3 induction depends on H. pylori T4SS. This expression pattern was reproduced both in human GECs and murine GEC (ImSt) infected with H. pylori. Besides using a mutant which lacks the whole cag PAI we also used an H. pylori mutant only devoid in the cagA gene to determine the role of this effector protein translocated by the T4SS in B7-H3 induction. Our study showed that induction of B7-H3 depends on the presence of CagA. PG, the other component translocated to GEC by T4SS may act as an inflammatory molecule and induces IL-8 production by GEC [14-16]. As these data showed complete dependence of H. pylori cag PAI but partial involvement of CagA on B7-H3 induction, we hypothesized PG, which is also translocated by T4SS, might also influence B7-H3 induction. The addition of PG fragment iEDAP which is recognized by NOD1 showed induction of B7-H3 both at the mRNA and protein levels. Kinetics data showed that B7-H3 is increased within 18-hrs of stimulation by PG fragments. This study also highlighted the involvement of p38 MAPK pathway in B7-H3 induction, which is known to be activated by both PG and CagA [14, 54]. Previously it was shown that PG can be modified to resist lysozyme and this mechanism helps H. pylori survival [55]. However, this is the first study showing role of PG in GEC modification and T cell regulation.
Cytokines play an important role in influencing the expression of different immune regulatory molecules. Since IL-17, IL-10 and TGF-β produced by Th17 and Treg cells have been shown to be present in increased amounts in H. pylori infected patient [34, 35, 37-39], we hypothesized that these cytokines may act in paracrine fashion to affect the induction of B7-H3 on GEC. Our data showed that stimulation of GECs by both Th17 cytokine (IL-17) and Treg cell cytokines (IL-10 and TGF-β) causes increased expression of B7-H3 molecules on GECs. Regulation of other B7 molecules by IL-10 and TGF-β have been shown previously. For instance, IL-10 was shown to inhibit B7 molecule expression in macrophages and B7-2 expression in DCs [56, 57]. Also, TGF-β has been found to inhibit B7-1 expression in APCs [58]. Another study showed IL-10 down-regulated B7-1 and B7-2 expression on Mycobacterium tuberculosis-infected monocytes to a greater extent than did TGF-β [59]. However, IL-10 and TGF-β did not show any additive or synergistic inhibition in their study, whereas, in this study, we found TGF-β is a better inducer of B7-H3 than is IL-10, and they have synergistic effects in B7-H3 induction.
To explore the contribution of B7-H3 to the development of T cell subsets in H. pylori infection, we initially determined what kind of T cell response ensues in mice infected with H. pylori strain in the presence or absence of a functional T4SS. To that end we measured different T cell associated transcription factors considered as master regulators for different CD4+ T cell subsets and cytokines produced by these cells in MLN harvested from H. pylori infected mice. Consistent with previous published data, we noted mixed populations of Th1 and Th2 cells in H. pylori infected mice [33]. Compared to SS1, PMSS1-infected mice had a lower induction of the Th1 and Th2 cell subsets. Additionally, Th17 and Treg cell data correlated with our previous findings, since H. pylori PMSS1 infection causes increased Treg cells and a lesser Th17 cell response when compared with findings in SS1 infected mice [41, 49]. Our lab has previously shown that H. pylori-mediated modulation of Th17 and Treg cell responses depends on altered expression of B7-H2 and B7-H1 molecules on GECs [41, 49]. Besides being a positive stimulator for T cell activation, B7-H3 has also been shown to play a role in Th2 development and to contribute to pathogenic Th2 cell development during asthma in a mouse model [26]. However, several studies also showed negative regulatory effects of B7-H3 in Th1 and Th2 immune responses [60]. A major question regarding the Th2 cell response observed in H. pylori-infected mice is whether or not this induction of Th2 is influenced by a B7-H3 molecule expression. To answer this question and investigate whether this response depends on the H. pylori strain, samples from H. pylori infected patients with either gastritis or tumor were collected and B7-H3 and GATA3 expression on those samples were compared with samples collected from healthy individuals. Interestingly, the samples collected from gastritis patients, and not from the gastric tumor patients, had increased B7-H3 and GATA3+ cells. Though this study showed a strong association between B7-H3 induction and Th2 development during H. pylori infection, further studies are required to determine the link between disease condition and B7-H3 expression. To further evaluate this finding, a GEC: T cell co-cultures were used, in which the GECs were pre-exposed to H. pylori strains which originated from gastritis, gastric cancer or peptic ulcer in the presence of anti-B7-H3-blocking antibody or control antibody. The flow cytometry data indicated the induction of Th2 cells and Th1/Th2 double-positive cells in the T cells co-cultured with H. pylori 51B (from a gastritis case) pre-treated cells, suggesting a shift of Th1 towards Th2 cells. Moreover, by using anti-B7-H3 blocking antibody, we showed that induction of Th2 depends on B7-H3.
Conclusion
In conclusion, this study revealed a novel mechanism that H. pylori uses to foster host chronic inflammation in the form of gastritis. This is an important finding which helps to better understand the interaction of H. pylori with GECs and how H. pylori manipulates the host T cell response. The relationship of H. pylori-mediated B7-H3 induction and disease conditions must be further defined.
Acknowledgements
This work was supported, in whole or in part, by Department of Defense Grant CA150375 to VER.
Abbreviations
GEC: Gastric epithelial cells;
T4SS: Type 4 secretion system;
PG: Peptidoglycan;
iEDAP: D-gamma-Glu-mDAP;
MALT: Mucosa-associated lymphoid tissue;
CagA: Cytotoxin associated gene A;
cag PAI: cag pathogenicity island;
Th: T-helper;
RSV: Respiratory syncytial virus;
Treg: T regulatory;
ATCC: American Type Culture Collection;
ImSt: Immortomouse stomach epithelium;
TSA: Tryptic soy agar;
SS1: H. pylori Sydney strain 1;
PM-SS1: Pre-mouse SS1;
WT: Wild type;
APC: Antigen presenting cells.
Article Info
Article Type
Research ArticlePublication history
Received: Sat 14, Sep 2019Accepted: Fri 27, Sep 2019
Published: Thu 10, Oct 2019
Copyright
© 2023 Victor E. Reyes. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited. Hosting by Science Repository.DOI: 10.31487/j.COR.2019.05.05
Figures & Tables



References
- Reyes VE, Peniche AG (2019) Helicobacter pylori Deregulates T and B Cell Signaling to Trigger Immune Evasion. In: Backert S. (eds) Molecular Mechanisms of Inflammation: Induction, Resolution and Escape by Helicobacter pylori. Curr Top Microbiol Immunol 421: 229-265. [Crossref]
- Malfertheiner P, Sipponen P, Naumann M, Moayyedi P, Megraud F et al. (2005) Helicobacter pylori eradication has the potential to prevent gastric cancer: a state-of-the-art critique. Am J Gastroentero 100:2 100-115. [Crossref]
- Uemura N, Okamoto S, Yamamoto S, Matsumura N, Yamaguchi S et al. (2001) Helicobacter pylori infection and the development of gastric cancer. N Eng J Med 345: 784-789. [Crossref]
- Blaser MJ, Perez-Perez GI, Kleanthous H, Cover TL, Peek RM et al. (1995) Infection with Helicobacter pylori strains possessing cagA is associated with an increased risk of developing adenocarcinoma of the stomach. Cancer Res 55: 2111-2115. [Crossref]
- Brenner H, Arndt V, Stegmaier C, Ziegler H, Rothenbacher D (2004) Is Helicobacter pylori infection a necessary condition for noncardia gastric cancer? Am J Epidemiol 159: 252-258. [Crossref]
- Odenbreit S, Puls J, Sedlmaier B, Gerland E, Fischer W et al. (2000) Translocation of Helicobacter pylori CagA into gastric epithelial cells by type IV secretion. Science 287: 1497-1500. [Crossref]
- Poppe M, Feller SM, Romer G, Wessler S (2007) Phosphorylation of Helicobacter pylori CagA by c-Abl leads to cell motility. Oncogene 26: 3462-3472. [Crossref]
- Stein M, Bagnoli F, Halenbeck R, Rappuoli R, Fantl WJ et al. (2002) c-Src/Lyn kinases activate Helicobacter pylori CagA through tyrosine phosphorylation of the EPIYA motifs. Mol Microbiol 43: 971-980. [Crossref]
- Tammer I, Brandt S, Hartig R, Konig W, Backert S (2007) Activation of Abl by Helicobacter pylori: a novel kinase for CagA and crucial mediator of host cell scattering. Gastroenterology 132: 1309-1319. [Crossref]
- Selbach M, Moese S, Hauck CR, Meyer TF, Backert S (2002) Src is the kinase of the Helicobacter pylori CagA protein in vitro and in vivo. J Biol Chem 277: 6775-6778. [Crossref]
- Backert S, Selbach M (2008) Role of type IV secretion in Helicobacter pylori pathogenesis. Cell Microbiol 10: 1573-1581. [Crossref]
- Bourzac KM, Guillemin K (2005) Helicobacter pylori-host cell interactions mediated by type IV secretion. Cell Microbiol 7: 911-919. [Crossref]
- Hatakeyama M (2004) Oncogenic mechanisms of the Helicobacter pylori CagA protein. Nat Rev Cancer 4: 688-694. [Crossref]
- Allison CC, Kufer TA, Kremmer E, Kaparakis M, Ferrero RL (2009) Helicobacter pylori induces MAPK phosphorylation and AP-1 activation via a NOD1-dependent mechanism. J Immunol 183: 8099-8109. [Crossref]
- Viala J, Chaput C, Boneca IG, Cardona A, Girardin SE et al. (2004) Nod1 responds to peptidoglycan delivered by the Helicobacter pylori cag pathogenicity island. Nat Immunol 5: 1166-1174. [Crossref]
- Watanabe T, Asano N, Kitani A, Fuss IJ, Chiba T et al. (2010) NOD1-mediated mucosal host defense against Helicobacter pylori. Int J Inflam 2010: 476482. [Crossref]
- Chapoval AI, Ni J, Lau JS, Wilcox RA, Flies DB et al. (2001). B7-H3: a costimulatory molecule for T cell activation and IFN-gamma production. Nat Immunol 2: 269-274. [Crossref]
- Steinberger P, Majdic O, Derdak SV, Pfistershammer K, Kirchberger S et al. (2004) Molecular characterization of human 4Ig-B7-H3, a member of the B7 family with four Ig-like domains. J Immunol 172: 2352-2359. [Crossref]
- Sun M, Richards S, Prasad DV, Mai XM, Rudensky A et al. (2002) Characterization of mouse and human B7-H3 genes. Journal of Immunology 168: 6294-6297. [Crossref]
- Zhang GB, Zhou H, Chen YJ, Ge Y, Xie F et al. (2005) Characterization and application of two novel monoclonal antibodies against 2IgB7-H3: expression analysis of 2IgB7-H3 on dendritic cells and tumor cells. Tissue Antigens 66: 83-92. [Crossref]
- Stanciu LA, Bellettato CM, Laza-Stanca V, Coyle AJ, Papi A et al. (2006) Expression of programmed death-1 ligand (PD-L) 1, PD-L2, B7-H3, and inducible costimulator ligand on human respiratory tract epithelial cells and regulation by respiratory syncytial virus and type 1 and 2 cytokines. J Infect Dis 193: 404-412. [Crossref]
- Suh WK, Gajewska BU, Okada H, Gronski MA, Bertram EM et al. (2003) The B7 family member B7-H3 preferentially down-regulates T helper type 1-mediated immune responses. Nat Immunol 4: 899-906. [Crossref]
- Castriconi R, Dondero A, Augugliaro R, Cantoni C, Carnemolla B et al. (2004) Identification of 4Ig-B7-H3 as a neuroblastoma-associated molecule that exerts a protective role from an NK cell-mediated lysis. Proc Natl Acad Sci USA 101: 12640-12645. [Crossref]
- Prasad DV, Nguyen T, Li Z, Yang Y, Duong J et al. (2004) Murine B7-H3 is a negative regulator of T cells. J Immunol 173: 2500-2506. [Crossref]
- Wang L, Fraser CC, Kikly K, Wells AD, Han R et al. (2005) B7-H3 promotes acute and chronic allograft rejection. Eur J Immunol 35: 428-438. [Crossref]
- Nagashima O, Harada N, Usui Y, Yamazaki T, Yagita H et al. (2008) B7-H3 contributes to the development of pathogenic Th2 cells in a murine model of asthma. J Immunol 181: 4062-4071. [Crossref]
- Arigami T, Narita N, Mizuno R, Nguyen L, Ye X et al. (2010) B7-h3 ligand expression by primary breast cancer and associated with regional nodal metastasis. Ann Surg 252: 1044-1051. [Crossref]
- Crispen PL, Sheinin Y, Roth TJ, Lohse CM, Kuntz SM et al. (2008) Tumor cell and tumor vasculature expression of B7-H3 predict survival in clear cell renal cell carcinoma. Clin Cancer Res 14: 5150-5157. [Crossref]
- Roth TJ, Sheinin Y, Lohse CM, Kuntz SM, Frigola X et al. (2007) B7-H3 ligand expression by prostate cancer: a novel marker of prognosis and potential target for therapy. Cancer Res 67: 7893-7900. [Crossref]
- Loos M, Hedderich DM, Ottenhausen M, Giese NA, Laschinger M et al. (2009) Expression of the costimulatory molecule B7-H3 is associated with prolonged survival in human pancreatic cancer. BMC Cancer 9: 463. [Crossref]
- Wu CP, Jiang JT, Tan M, Zhu YB, Ji M et al. (2006) Relationship between co-stimulatory molecule B7-H3 expression and gastric carcinoma histology and prognosis. World J Gastroenterol 12: 457-459. [Crossref]
- Arigami T, Uenosono Y, Hirata M, Yanagita S, Ishigami S et al. (2011) B7-H3 expression in gastric cancer: a novel molecular blood marker for detecting circulating tumor cells. Cancer Sci 102: 1019-1024. [Crossref]
- Goll R, Gruber F, Olsen T, Cui G, Raschpichler G et al. (2007) Helicobacter pylori stimulates a mixed adaptive immune response with a strong T-regulatory component in human gastric mucosa. Helicobacter 12: 185-192. [Crossref]
- Cheng HH, Tseng GY, Yang HB, Wang HJ, Lin HJ et al. (2012) Increased numbers of Foxp3-positive regulatory T cells in gastritis, peptic ulcer and gastric adenocarcinoma. World J Gastroenterol 18: 34-43. [Crossref]
- Lundgren A, Suri-Payer E, Enarsson K, Svennerholm AM, Lundin BS (2003) Helicobacter pylori-specific CD4+ CD25high regulatory T cells suppress memory T-cell responses to H. pylori in infected individuals. Infect Immun 71: 1755-1762. [Crossref]
- Kabir S (2011) The role of interleukin-17 in the Helicobacter pylori induced infection and immunity. Helicobacter 16: 1-8. [Crossref]
- Algood HM, Gallo-Romero J, Wilson KT, Peek RM Jr, Cover TL (2007) Host response to Helicobacter pylori infection before initiation of the adaptive immune response. FEMS Immunol Med Microbiol 51: 577-586. [Crossref]
- Caruso R, Fina D, Paoluzi OA, Del Vecchio Blanco G, Stolfi C et al. (2008) IL-23-mediated regulation of IL-17 production in Helicobacter pylori-infected gastric mucosa. Eur J Immunol 38: 470-478. [Crossref]
- Luzza F, Parrello T, Monteleone G, Sebkova L, Romano M et al. (2000) Up-regulation of IL-17 is associated with bioactive IL-8 expression in Helicobacter pylori-infected human gastric mucosa. J Immunol 165: 5332-5337. [Crossref]
- Das S, Suarez G, Beswick EJ, Sierra JC, Graham DY et al. (2006) Expression of B7-H1 on gastric epithelial cells: its potential role in regulating T cells during Helicobacter pylori infection. J Immunol 176: 3000-3009. [Crossref]
- Lina TT, Pinchuk IV, House J, Yamaoka Y, Graham DY et al. (2013) CagA-dependent downregulation of B7-H2 expression on gastric mucosa and inhibition of Th17 responses during Helicobacter pylori infection. J Immunol 191: 3838-3846. [Crossref]
- Beswick EJ, Pinchuk IV, Das S, Powell DW, Reyes VE (2007) Expression of the programmed death ligand 1, B7-H1, on gastric epithelial cells after Helicobacter pylori exposure promotes development of CD4+ CD25+ FoxP3+ regulatory T cells. Infect Immun 75: 4334-4341. [Crossref]
- Whitehead RH, Robinson PS (2009) Establishment of conditionally immortalized epithelial cell lines from the intestinal tissue of adult normal and transgenic mice. Am J Physiol Gastrointest Liver Physiol 296: G455-G460. [Crossref]
- Beswick EJ, Pinchuk IV, Suarez G, Sierra JC, Reyes VE (2006) Helicobacter pylori CagA-dependent macrophage migration inhibitory factor produced by gastric epithelial cells binds to CD74 and stimulates procarcinogenic events. J Immunol 176: 6794-6801. [Crossref]
- Crowe SE, Alvarez L, Dytoc M, Hunt RH, Muller M et al. (1995) Expression of interleukin 8 and CD54 by human gastric epithelium after Helicobacter pylori infection in vitro. Gastroenterology 108: 65-74. [Crossref]
- Bjorkholm BM, Guruge JL, Oh JD, Syder AJ, Salama N et al. (2002) Colonization of germ-free transgenic mice with genotyped Helicobacter pylori strains from a case-control study of gastric cancer reveals a correlation between host responses and HsdS components of type I restriction-modification systems. J Biol Chem 277: 34191-34197. [Crossref]
- Arnold IC, Lee JY, Amieva MR, Roers A, Flavell RA et al. (2011) Tolerance rather than immunity protects from Helicobacter pylori-induced gastric preneoplasia. Gastroenterology 140: 199-209. [Crossref]
- Beswick EJ, Pinchuk IV, Earley RB, Schmitt DA, Reyes VE (2011) Role of gastric epithelial cell-derived transforming growth factor beta in reduced CD4+ T cell proliferation and development of regulatory T cells during Helicobacter pylori infection. Infect Immun 79: 2737-2745. [Crossref]
- Lina TT, Alzahrani S, House J, Yamaoka Y, Sharpe AH et al. (2015) Helicobacter pylori cag pathogenicity island's role in B7-H1 induction and immune evasion. PLoS One 10(3): e0121841. [Crossref]
- Higashi H, Nakaya A, Tsutsumi R, Yokoyama K, Fujii Y et al. (2004) Helicobacter pylori CagA induces Ras-independent morphogenetic response through SHP-2 recruitment and activation. J Biol Chem 279: 17205-17216. [Crossref]
- Fujimura T, Ring S, Umansky V, Mahnke K, Enk AH (2012) Regulatory T cells stimulate B7-H1 expression in myeloid-derived suppressor cells in ret melanomas. J Invest Dermatol 132: 1239-1246. [Crossref]
- Franco AT, Israel DA, Washington MK, Krishna U, Fox JG et al. (2005) Activation of beta-catenin by carcinogenic Helicobacter pylori. Proc Natl Acad Sci USA 102: 10646-10651. [Crossref]
- Mimuro H, Suzuki T, Nagai S, Rieder G, Suzuki M et al. (2007) Helicobacter pylori dampens gut epithelial self-renewal by inhibiting apoptosis, a bacterial strategy to enhance colonization of the stomach. Cell Host Microbe 2: 250-263. [Crossref]
- Keates S, Keates AC, Warny M, Peek RM Jr, Murray PG et al. (1999) Differential activation of mitogen-activated protein kinases in AGS gastric epithelial cells by cag+ and cag- Helicobacter pylori. J Immunol 163: 5552-5559. [Crossref]
- Wang G, Lo LF, Forsberg LS, Maier RJ (2012) Helicobacter pylori peptidoglycan modifications confer lysozyme resistance and contribute to survival in the host. MBio 23: e00409-e00412. [Crossref]
- Ding L, Linsley PS, Huang LY, Germain RN, Shevach EM (1993) IL-10 inhibits macrophage costimulatory activity by selectively inhibiting the up-regulation of B7 expression. J Immunol 151: 1224-1234. [Crossref]
- Buelens C, Willems F, Delvaux A, Pierard G, Delville JP et al. (1995) Interleukin-10 differentially regulates B7-1 (CD80) and B7-2 (CD86) expression on human peripheral blood dendritic cells. Eur J Immunol 25: 2668-2672. [Crossref]
- Xu H, Silver PB, Tarrant TK, Chan CC, Caspi RR (2003) Tgf-beta inhibits activation and uveitogenicity of primary but not of fully polarized retinal antigen-specific memory-effector T cells. Invest Ophthalmol Vis Sci 44: 4805-4812. [Crossref]
- Rojas RE, Balaji KN, Subramanian A, Boom WH (1999) Regulation of human CD4(+) alphabeta T-cell-receptor-positive (TCR(+)) and gammadelta TCR(+) T-cell responses to Mycobacterium tuberculosis by interleukin-10 and transforming growth factor beta. Infect Immun 67: 6461-6472. [Crossref]
- Fukushima A, Sumi T, Fukuda K, Kumagai N, Nishida T et al. (2007) B7-H3 regulates the development of experimental allergic conjunctivitis in mice. Immunol Lett 113: 52-57. [Crossref]
- Björkholm BM, Guruge JL, Oh JD, Syder AJ, Salama N et al. (2002) Colonization of germ-free transgenic mice with genotyped Helicobacter pylori strains from a case-control study of gastric cancer reveals a correlation between host responses and HsdS components of type I restriction-modification systems. J Biol Chem 277: 34191-34197. [Crossref]
- Ohnishi N (2008) Proc Natl Acad Sci U S A. 105: 1003-1008.
- Suarez G, Romero-Gallo J, Pizauelo MB, Wang G, Maier RJ et al. (2015) Modification of Helicobacter pylori peptidoglycan enhances NOD1 activation and promotes cancer of the stomach. Cancer Res 75: 1749-1759. [Crossref]