Journals
Genes and proteins associated with chemotherapeutic resistance in breast cancer
A B S T R A C T
Breast cancer is the main type of cancer that affects women in the world. Treatment with chemotherapeutic agents for this type of cancer depends to a large extent on the phenotypic characteristics that appear in the tumor, such as some receptors.
The treatment of breast cancer is complex since not all patients respond adequately to it and this is partly due to mechanisms of resistance to treatment. The resistance either acquired or intrinsic to chemotherapeutics against breast cancer is related to multiple genes and proteins that are actively involved in the development of resistance mechanisms, which may be inhibiting the binding of the drug to the target receptor, inhibiting the gene expression of the receptors, activating signaling pathways, inducing the action of transporters that expel the drug from the cell, etc.
As a result of this review, the mechanisms of chemotherapeutic resistance in breast cancer derived from genes and proteins related to say disease are described, including genes related to different types of breast cancer, as well as with different therapeutic strategies.
These genes and their products of expression include a wide range of interactions and activations of signalling pathways that trigger a poor response to treatment, which translates into a worse prognosis, higher risk of prevalence and death, favouring breast cancer to be the main cause of cancer death in women.
Objective: Conduct an updated review of genes and proteins, that have been associated with conferring resistance to the different chemotherapeutic agents used for the treatment of breast cancer, including the mechanism of resistance and the type of cancer in which it is generated.
Method: The search for indexed articles in the PubMed (https://www.ncbi.nlm.nih.gov/pubmed/) and ScienceDirect (http://www.sciencedirect.com) databases was performed, using the words “Breast cancer resistance” as a search criterion, articles published in the period 2015-2017 were selected.
K E Y W O R D S
Breast cancer, resistance, genes, proteins, chemotherapeutics, mechanisms
I N T R O D U C T I O N
Breast cancer is the predominant type of cancer in women with 25% of new cases and 15% of deaths caused by oncological diseases in the world [1]. Breast cancer is an extremely diverse and complex disease, where there is a marked tumour heterogeneity among patients, with specific subtypes of breast cancer associated with different prognoses. Differentiating breast tumour types is a key component of the clinical management process to ensure that patients receive the most appropriate type of treatment [2].
The treatment of breast cancer includes different types of therapies that can be local or systemic. Local therapy includes surgery and radiotherapy, while chemotherapy, hormonal therapy and targeted therapy are among the systemic therapies [3].
The choice of treatment for breast cancer depends on the pathological characteristics that occur in each patient, such as the stage of the tumour, the state of the hormone receptors presents in the tumour, as well as the rate of recurrence, for example, in patients’ low recurrence, score hormonal therapy is recommended, while chemotherapy is mandatory for patients with high recurrence score. For patients with an intermediate recurrence score, the systemic therapy chosen takes into account risk factors such as age, tumour size, degree of tumour differentiation, extension of lymph node invasion, etc. [3, 4].
Avoiding resistance to therapeutic drugs is a critical point for researchers and clinicians, since it is desired to achieve successful therapy in patients with cancer. Drug resistance can be classified as intrinsic resistance or acquired resistance. Intrinsic resistance refers to patients who develop pre-existing factors and mediators of resistance before receiving chemotherapy. Acquired resistance on the other hand focuses on patients who were initially sensitive to chemotherapy but developed resistance during treatment [5].
There are different mechanisms that can lead to resistance to chemotherapeutic drugs such as inactivation of the drug, alteration of the site of action of the drug, drug efflux, repair of DNA damage, inhibition of cell death and epithelial–mesenchymal transition (EMT) [5].
Within these mechanisms influence different genes and proteins, as shown in Table 1, which respond against the treatment and therefore there is ineffectiveness of it, what leads to patients not improving their condition, below, certain genes and proteins associated with resistance to treatment against breast cancer are referred to. Genes and proteins associated with the resistance of breast cancer treatment:
Table 1: Genes and proteins associated with chemotherapeutic resistance in different types of breast cancer
|
Gene/Protein resistance effectors |
Abbreviation |
Chemotherapeutics affected by resistance |
Type of cancer where resistance is presented |
|
Fosfoserin aminotransferase-1
|
PSAT-1 |
Tamoxifen |
Luminal A and B (RE+, RP+) |
|
SOX9 embryonic transcription factor
|
SOX9 |
Tamoxifen |
Luminal A and B (RE+, RP+) |
|
Initiation complex of the eIF4F translation |
eIF4F |
Tamoxifen |
Luminal A and B (RE+, RP+) |
|
Leptin |
LEP |
Tamoxifen |
Luminal A and B (RE+, RP+) |
|
Mucin-1 protein |
MUC-1 |
Bortezomib, trastuzumab, tamoxifen |
Luminal A and B (RE+, RP+), positive Her-2 |
|
Glutathione S-transferase P1 |
GSTP-1 |
Anthracyclines and taxanes such as: adriamycin, daunomycin, epirubicin, docetaxel, paclitaxel, cyclophosphamide and doxorubicin |
Breast carcinoma in general |
|
β-glucosidase |
GBA |
Paclitaxel |
Breast carcinoma in general |
|
ESR-1 Gene |
ESR-1 |
Aromatase inhibitors such as: letrozole, anastrozole, exemestane |
Luminal A and B (RE+, RP+) in postmenopausal women. |
|
miRNA |
miRNA221/222, miRNA-342-3p, miRNA-873 |
Tamoxifen |
Luminal A and B (RE+, RP+) |
|
Interleukin 33 |
IL-33 |
Tamoxifen |
Luminal A and B (RE+, RP+) |
|
Factor E2F8 |
E2F8 |
Cisplatin |
Breast carcinoma in general |
|
Gene/Protein resistance effectors |
Abbreviation |
Chemotherapeutics affected by resistance |
Type of cancer where resistance is presented |
|
Mucin 4 |
MUC4 |
Trastuzumab |
Positive Her-2 |
|
ATP binding cassette transport protein G2 |
ABCG2 |
Mitoxantrone and camptothecin, topotecan and irinotecan derivatives |
Metastatic breast cancer |
|
Survivin |
BIRC-5 |
Taxans, kinesin inhibitors and mTOR inhibitors |
Breast carcinoma in general |
|
Brachyury protein / SIRT1 gene |
T/SIRT1 |
Tamoxifen |
Luminal A and B (RE+, RP+) |
|
Membrane cell adhesion molecule |
MCAM CD146 |
Tamoxifen |
Luminal A and B (RE+, RP+) |
|
Cyclin E |
CCNE-1 |
Aromatase inhibitors such as: letrozole, anastrozole, exemestane |
Luminal A and B (RE+, RP+)in postmenopausal women. |
|
Nuclear factor enhancer of the kappa light chains of activated B cells |
NF-κB |
RTK inhibitors such as trastuzumab, bevacizumab or imatinib |
Breast cancer triple negative marker |
|
Krüppel-like factor 4 |
KLF4 |
Tamoxifen |
Luminal A and B (RE+, RP+) |
|
Cancerous protein phosphatase 2A inhibitor |
CIP2A |
Tamoxifen |
Luminal A and B (RE+, RP+) |
|
p95-HER2 |
p95-HER2 |
Trastuzumab T-DM1 |
Positive Her-2 |
|
Exostosin 1 |
EXT1 |
Doxorubicin |
Breast carcinoma in general |
|
Cytochrome P450 2D6 |
CYP2D6 |
Tamoxifen |
Luminal A and B (RE+, RP+) |
Phosphoserine aminotransferase 1 (PSAT1)
PSTA1 is an amino-transferase enzyme involved in the conversion of phospho-pyruvate, which is derived from the oxidation of 3-phosphoglycerate, to phosphoserine [6]. Phosphoserine is then converted to serine by the enzyme phosphoserine phosphatase and is further converted to glycine to feed the nucleotide biosynthesis pathway. Along with this, it has been shown that the serine biosynthetic pathway is a critical factor in tumorigenesis and resistance to breast cancer therapy [6].
PSAT1 has been positively associated with the expression of some signaling pathways such as JAK / STAT and the activation of some receptors for cytokines such as IL4R and IL21R [6]. The IL4R receptor has been associated with the metabolic reprogramming of tumour cells in favour of an increase in glucose uptake and glutamine metabolism, whereas the expression of IL21R has been linked to the proliferation of breast cancer cells in the MDA-MB 231 cell line, in addition the binding of IL21 to its receptor promotes the activation of several signalling cascades, such as the JAK-STAT pathway, with differential effects related to tissue expression [6].
The JAK-STAT signalling pathway induces metabolic interruptions in tumour cells and facilitates the progression of the cell cycle by promoting the activation of cyclin-dependent kinases, so that the enrichment of PSAT1 is able to increase the expression of the pathway and because the JAK-STAT pathways and cytokines are directly related to inflammation, they contribute to the growth and spread of the tumour [6]. Therefore, an increase in the presence of PSAT1 protein and mRNA levels is significantly associated with the poor outcome of treatment with tamoxifen in breast cancer [6].
Embryonic transcription factor SOX9(PSAT1)
The estrogen receptor (ER) drives the growth of most luminal breast cancers, which is the main goal of endocrine therapy. Although ER blockade with drugs such as tamoxifen is very effective, one of the main clinical limitations is the development of endocrine resistance, this can be seen in Figure 1, which shows the action of estrogen and tamoxifen on the estrogen receptor and some molecules involved in resistance to this therapy [7].
It has been identified that treatment with tamoxifen promotes a complex called RUNX2-ER, which facilitates the positive regulation of the embryonic transcription factor SOX9, which is a transcription factor that regulates stem and progenitor cells in adult tissues. Recently it has been shown that they have a key role in the development of metastases in triple negative and HER2-positive breast cancer. It has been found that SOX9 is also expressed in breast cancers positive to estrogen receptor alpha, where it increases with progression, and plays an important role in endocrine resistance [7].
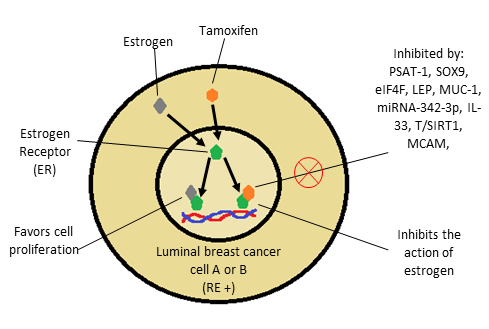
Figure 1: Action of estrogen and tamoxifen on the estrogen receptor. Molecules that inhibit the effect of tamoxifen, generating resistance
It was shown that SOX9 is regulated in tamoxifen resistant cells (TAMR) by the ER α-RUNX2 complex, the proliferation of TAMR cells is dependent on SOX9, and this regulation is sufficient to cause resistance to estrogen deprivation and decreased sensitivity to treatment with tamoxifen [7].
Initiation complex of the eIF4F translation
The translation initiation complex eIF4F consists of an RNA helicase (eIF4A), a scaffold protein (eIF4G) and the eukaryotic translation initiation factor of the 4E cap binding protein (eIF4E), which is the limiting component of the complex. Evidence from multiple experiments suggests that malignancies driven by different oncogenic pathways converge and depend on the hyperactivation of the eIF4F translation machinery. It has been identified that in breast cancer cells resistant to tamoxifen there is a hyperactivation of the eIF4F pathway, which prevents the correct function of this therapeutic agent in the control of breast cancer [8].
Leptin
Leptin, the product of the Ob gene, is a cytokine released by adipose tissue. It acts as a multifunctional peptide hormone that can modulate broad biological processes, including appetite regulation, reproductive function, immune response, angiogenesis, etc. Leptin exerts its function by binding to its receptor (ObR), where the long ObRb isoform contains the full-length intracellular domain and has a total signalling potential [9].
Leptin plays an important role in the progression of breast cancer and interferes with the action of tamoxifen, since it interferes with the action of this treatment in breast cancer cells through the activation of signalling pathways ERK1 / 2 and STAT3 [9].
Recent studies have shown that the signal transduction pathway ERK1 / 2 is important in the signalling of leptin to promote the growth, proliferation and migration of MCF-7 cells [9]. Overexpression of leptin-mediated cyclin D1 may also be associated with leptin-induced tamoxifen resistance in breast cancer, since leptin was found to induce a robust increase in cyclin D1 expression mediated by activation of STAT3 [9].
The overexpression of cyclin D1 is an alteration that participates in the interruption of the cell cycle in human tumours and serves as a critical interface between hormonal signalling and tumorigenesis, so overexpression of cyclin D1 correlates with poor prognosis in ER-positive breast cancer and affects the response of tamoxifen in breast cancer cell lines [9].
Mucin1 protein (MUC1)
The MUC1 protein is a glycosylated transmembrane protein of the epithelium, whose main function is to protect cells from the stresses induced by the external environment [10]. During the stress condition, MUC1 generates the transduction of survival signals into the interior of the cell, essential to maintain its functionality [10].
Recent studies have shown that a domain of this protein known as cytoplasmic domain MUC1 (MUC1-C) accelerates the development of resistance to several cancer therapeutics including bortezomib, trastuzumab and tamoxifen [10]. MUC1-C promotes the development of resistant mechanisms through signalling in the cell membrane and activation of signal transduction pathways [11].
The overexpression of MUC1-C in breast cancer cells triggers resistance to tamoxifen, by means of the following mechanism, where the phosphorylation of the cytoplasmic tail is able to interact with the ERα nuclear estrogen receptor. In the case of resistance to anticancer therapeutic agents, ER leads to sustained downstream signalling. Even in the presence of therapeutic agents, the aberrant activation of the PI3K / AKT pathway in the progression of the breast tumour leads to the inhibition of GSK3 and the activation of Bcl-2, thus blocking the apoptotic process [11].
Glutathione S-transferase P1 (GSTP1)
Glutathione S-transferase P1 (GSTP1), an enzyme that belongs to the family of metabolic phase II enzymes, is fundamental in the detoxification of potentially harmful chemical compounds by conjugating these compounds with glutathione [12]. Studies have identified that an overexpression of the enzyme GSTP1 participates as an agent that promotes resistance to chemotherapy in patients with breast cancer, especially in the treatments with adriamycin, daunomycin, epirubicin, docetaxel, paclitaxel, cyclophosphamide and doxorubicin [12].
There are different mechanisms that explain the resistance caused by this enzyme, one of them corresponds to an increase in the excretion of drugs because they are conjugated with glutathione, which gives them greater polarity and water solubility [12]. Another mechanism involves the presence of exosomes containing high levels of GSTP1 that appear to be essential for conferring resistance to anticancer drugs and are proposed as biomarkers of drug resistance derived from anthracyclines and taxanes [12].
β-glucosidase (GBA)
β-glucosidase is a lysosomal enzyme that cleaves the glucosidic bond of glucosylceramide to generate ceramide and release glucose. It has been found to be involved in some pathologies such as Gaucher's disease and Parkinson's disease because defects in their activity lead to the accumulation of lipid substrates [13].
This enzyme has been linked to breast cancer, since an increase in the activity of β-glucosidase has been observed in breast cancer cells. By analysing different breast cancer cell lines, it was concluded that the high activity of β-glucosidase is a common phenomenon in breast cancer regardless of the different cell origins and genetic background [13].
It has been found that the activation of β-glucosidase results in the promotion of growth in the cells of the breast and the stimulation of the oncogenic pathway by increasing the levels of phosphorylation of the essential molecules involved in the PI3K / Akt / mTOR pathway [13]. Treatment with chemotherapy does not affect the activity of β-glucosidase, suggesting that this enzyme is not involved in the response of breast cancer cells to chemotherapy. However, the inhibition of β-glucosidase significantly increased the efficacy of paclitaxel. These data clearly demonstrate that the inhibition of β-glucosidase is capable of sensitizing breast cancer cells to chemotherapy and is an important factor in avoiding resistance to therapy in breast cancer [13].
ESR-1 gene
Recent studies have shown changes in subclonal structure through treatment interventions using cytotoxic and endocrine therapy. Recently, several genomic mechanisms of resistance to various therapies have been discovered, with the mutations in ESR1, the gene that codes for the estrogen receptor, being the most described and studied in the context of endocrine resistance [14]. Mutations are grouped in the ligand-binding domain of ESR1 and activate the estrogen receptor in the absence of the estrogen ligand. These mutations have been detected almost exclusively in the context of acquired endocrine therapy, typically with aromatase inhibitors, and more frequently where sensitivity to endocrine therapy was previously observed, suggesting that these mutations are associated with acquired endocrine resistance and not with primary resistance to treatment [14].
miRNA
Endocrine therapies include selective modulators of estrogen receptors such as tamoxifen and aromatase inhibitors such as anastrozole and letrozole. In the case of the alpha estrogen receptor, mutations have been identified that result in ligand-independent receptor activation and changes in microRNA expression [15].
The miRNAs are small non-coding RNAs with an average size of 22 nucleotides that post-transcriptionally regulate gene expression by binding to the 30 untranslated region (30UTR) of miRNAs to suppress translation or promote mRNA degradation [15]. These miRNAs have specific expression profiles for cancer, since they have been implicated in the regulation of breast cancer characteristics, including cell proliferation, cell death, apoptosis, the immune response, the energy cell cycle, the metabolism, replicative immortality, senescence, invasion and metastasis [15, 16].
An example of this is miRNA-222, which reduces the expression of FOXO1 which is one of the members of the O subfamily (FOXO) and which acts as a tumor suppressor, regulating many biological processes that involve the apoptotic response, the regulation of the cell cycle, cell differentiation and cellular metabolism [17].
Some examples of miRNAs, which are actively involved in resistance to endocrine treatment of breast cancer are. miRNA-221/222, miRNA-342-3 p, miRNA-873 who negatively regulate the expression of the estrogen receptor alpha, which provides fewer sites of action for endocrine therapies and, therefore, in combination with other deregulated miRNAs, serves as a mechanism for resistance [15].
Another mechanism that associates resistance to treatment in breast cancer with miRNAs is that involving miRNA-342-3p, where the loss of said miRNA was associated with a concomitant loss in the expression of estrogen receptor alpha and resulted in resistance to tamoxifen therapy. It has also been determined that forced overexpression of miR-342-3p sensitized MCF7 breast cancer cells to apoptosis induced by tamoxifen [16].
Interleukin 33 (IL-33)
IL-33, belongs to the family of cytokines 1 (IL-1), is a double-function protein that acts as an alarming extracellular cytokine and as an intracellular nuclear factor. IL-33 is constitutively expressed in epithelial barrier tissues and lymphoid organs, maintaining the barrier function under normal conditions, and is released as a warning signal of cell damage or stress. In patients with breast cancer, serum IL-33 is significantly higher and correlates with levels of VEGF, MMP-11 and PDGF-C that indicate a poor prognosis [18]. The expression of IL-33 in tumor tissues participates in the prediction of aggressive phenotype of cancer cells and increased involvement of lymph node involvement in patients with breast cancer, Likewise, signaling of the IL-33 / ST2 complex promotes the progression of breast cancer through the support of inflammation and tumorigenesis, facilitating the expression of pro-angiogenic VEGF and attenuating tumor necrosis [18].
An additional mechanism states that IL-33 contributes to tumor immune evasion by promoting the expansion and immunosuppressive function of myeloid suppressor cells. These findings consider IL-33 as a pro-cancer cytokine important in the development of breast cancer [18]. The expression of IL-33 in tumor cells predicts a higher activity of ALDH, a greater expression of SOX2, a decrease in the expression of the estrogen receptor and therefore favors resistance to tamoxifen [18].
E2F8 factor
The E2F8 factor acts as a transcription activator that positively regulates the expression of CCNE1 and CCNE2 through interacting directly with their respective gene promoter, thereby promoting the proliferation of cancer cells [19]. Cisplatin is one of the most successful and commonly used chemotherapeutic drugs; however, tumors initially sensitive to chemotherapeutic agents usually develop resistance to these drugs, which is the main cause of relapse, metastasis and, finally, death related to cancer [19].
The treatment with cisplatin leads directly to the activation of the control point in the G2 / M phase, which subsequently leads to cellular apoptosis to eliminate the damaged cells. This activation of the control point occurs predominantly in the late phases S and G2, while the cells in phase M are reported to be hypersensitive to cisplatin. It has been shown that overexpression of E2F8 factor prevents cisplatin-induced cellular apoptosis in MCF-7 cells by shortening the arrest of G2 / M cell phases [19].
Brachyury protein / SIRT1 gene
The brachyury protein is a necessary transcription factor for the specification of the mesoderm during the development of the embryo, but it is absent in adults recently, this protein has been associated with the tumor aggressiveness in several types of tumors such as breast cancer [20].
The brachyury protein is found to be expressed in samples of breast cancer tumors and cell lines; which is associated with an increased risk of recurrence; and affects tumorigenesis of breast cancer, including proliferation, migration, tumor dissemination and increased resistance to chemotherapy [20]. SIRT 1 is an oncogene that is found to be expressed in malignant tumors of the prostate, breast, pancreas and liver. It plays a critical role in multiple aspects of resistance to cancer chemotherapy and promotes the survival of cancer cells [20].
It has been found that overexpression of the brachyury protein reduces the expression of SIRT1. Silencing of SIRT1 results in resistance to tamoxifen. This suggests that over expression of the brachyury protein induces tamoxifen resistance through the inhibition of SIRT1 expression [20].
MCAM
MCAM, also called CD146 or MUC18, was identified for the first time in malignant melanoma and was shown to be a key oncogene driving the progression of melanoma and metastasis. The new clinical evidence indicates that the presence of MCAM confers a poor prognosis in patients with breast cancer [21]. It has been shown that MCAM is over expressed in cancer of basal type or with over expression of Her-2, but not in luminal cancers, likewise an inverse correlation has been found between the expression of MCAM and the expression of ERa in cell lines of breast cancer [21].
Since the effects of tamoxifen are mediated primarily by its binding to ER, loss of ER expression may confer resistance to treatment. It is suggested that the mechanism by which MCAM suppresses the expression of ERa is given transcription level by the regulation of Slug expression, since it is suggested that Slug binds directly to box E in the region of the ERa promoter to control the activation and function of ERa [21].
Cyclin E
Cyclin E, a key regulatory protein that controls the transition between the G1 phase and the S phase in cells, is post translationally modified by proteolytic cleavage to generate cyclin E low molecular weight isoforms (LMW-E). These isoforms of cyclin E have been identified in different types of cancer such as: breast cancer, ovarian cancer, melanoma, colorectal cancer, lung cancer and renal cell carcinoma and are associated with a decrease in patient survival [22].
It has been identified that LMW-E are able to generate resistance to aromatase inhibitors (anastrozole, letrozole, and exemestane) that interfere with the enzymatic conversion of androgens to estrogens, but not with the biosynthesis of ovarian estrogens, making them effective only in postmenopausal patients [22]. The mechanism by which the expression of LMW-E mediates the resistance to aromatase inhibitor therapy is an alteration of the cell cycle of these cells in the transition from G1 to S. Specifically, the expression of LMW-E induces a greater number of arrested cells in the S phase, and due to the LMW-E mediated phosphorylation of pRb, cells can no longer be stopped in the G1 phase of the cell cycle [22].
Most endocrine therapies mediate their cytostatic effects causing the arrest of cells in the G1 phase of the cell cycle, thus inhibiting cell proliferation. Changes in the G1-S-S transition can be considered as an early event in the endocrine resistance pathway [22].
NF-κB
Triple-negative breast cancer (TNBC) is defined as a specific subtype of epithelial breast tumors, negative for expressing the estrogen receptor (ER), the progesterone receptor (PR) and the HER2. Contrary to other types of tumors, a special chemotherapy protocol has not been established to treat TNBC [23].
The absence of the ER / PR / HER2 overexpression pattern has made endocrine therapy with tamoxifen, aromatase inhibitors and therapies directed by HER2 useless for this type of cancer. In addition, TNBC is resistant to anthracyclines and taxanes, making the number of powerful therapeutic agents more restricted [23].
Recent studies have shown that tyrosine kinase (RTK) receptors are overexpressed in TNBC. Multiple members of these cell surface receptors have been identified and have been shown to play a key role in the regulation of essential cellular processes, including proliferation, differentiation, cell survival, migration and cell cycle control [23].
The epidermal growth factor receptor (EGFR), the fibroblast growth factor receptor (FGFR), the platelet-derived growth factor receptor (PDGFR), the vascular endothelial growth factor receptor (VEGFR) and the receptor insulin-like growth factor (IGF1-R) are the most important RTK presented in TNBC cells. However, chemotherapy with its selective inhibitors, which include trastuzumab, bevacizumab or imatinib, does not show significant improvement or decrease in the rate of progression of this type of cells [23].
Nuclear factor B (NF-κB) is involved in the expression of pro-inflammatory proteins including cytokines, chemokines and adhesion molecules. Tumors can increase the activity of NF-κB by increasing the release of stromal cell cytokines and fibroblasts in the tumor microenvironment. Overexpression of NF-κB has been found in the TNBC and since it is the point of convergence of several signaling pathways transduced by RTK, it is suggested that overexpression of NF-κB can be considered as one of the main reasons why RTK inhibitors do not induce the desired responses in clinical trials [23].
The activation of NF-κB leads to the expression of several pro-inflammatory cytokines that generate a favorable microenvironment for tumor propagation in conjunction with growth factors, which lead to the formation of new mutations in cancer cells and the further development of resistance to agents of chemotherapy with RTK inhibitors. Within these pro-inflammatory cytokines, interleukin 6 and interleukin 8 are included, who develop crosstalk’s with RTKs and NF-κB. This mechanism may be the response to resistance to chemotherapy despite the administration of RTK inhibitors [23].
An additional mechanism is based on the fact that some external stimulators such as reactive oxygen species (ROS) and hypoxia-induced factor (HIF) can promote the activation of NF-κB. These stimulators have been cataloged as factors inducing chemo-resistance, which leads to the conclusion that the overexpression of NF-κB participates in the development of resistance to RTK inhibitors [23].
Survivin
Survivin is a protein that acts as an inhibitor of apoptosis, by inhibiting caspases 3 and 7. It also inhibits cell death by interfering with the processing of caspase-9, the main inhibitor in the intrinsic pathway of apoptosis. It has been determined that the expression of this protein is minimal in normal tissues and is expressed in a large number of malignant tumors, where their levels of expression correlate positively with a more aggressive condition and a poor clinical outcome, which has made it a target for diagnosis, prognosis and for cancer therapies [24]. The overexpression of survivin in cancer can overcome the control points of the cell cycle to facilitate the aberrant progression of transformed cells through mitosis, in addition it has been demonstrated that the expression of survivin induces transcriptional changes in the tissue microenvironment promoting tumorigenesis [24].
The positive regulation of survivin contributes to establish resistance to taxanes, kinesin inhibitors and mTOR inhibitors in breast cancer cells, this has been demonstrated by performing survivin inhibition assays in samples resistant to chemotherapy and inducing the anticancer activity of these drugs, and it was found that by suppressing the activity of survivin, chemotherapy provides good results [25].
These data reinforce the hypothesis that survivin is a mediator of resistance to chemotherapy and it is suggested that the increase of this protein in cancer tissues is an unfavorable prognostic marker, which is associated with an increased risk of recurrence [25].
Mucin 4 (MUC4)
The mucin family (MUC) consists of glycosylated transmembrane proteins. Overexpression of some members of this family such as MUC1, MUC4 and MUC16, has been found in several human carcinomas, including breast cancer [26]. The MUC4 protein has three domains (NIDO, AMOP and VWD) that have not been found in any other protein belonging to the mucin family and functions as a ligand for the tyrosine kinase of the ErbB2 receptor [26].
Overexpression of MUC4 favors tumor development and cancer progression. Said overexpression has been identified in 30% to 95% of all types of breast cancer, and it is related to lymph node metastases and vascular tumor emboli [27].
It has been found that the MUC4 protein participates actively in resistance to treatment with trastuzumab, which in addition to being used in standard chemotherapy has been shown to improve the outcome of treatment of the early stage of breast cancer, as well as in cancer of HER2-positive metastatic breast. However, despite this success, responses to trastuzumab have been hampered by several resistance mechanisms, including overexpression of MUC4 [27]. The mechanism by which MUC4 participates in resistance to trastuzumab, is that this large glycoprotein can considerably hinder the accessibility and, therefore, the binding of trastuzumab to the ectodomain HER2 through the formation of a ligand / modulator complex, which impairs effective treatment with trastuzumab [27]..
Polymorphism of the factor (V) of Leiden (F5 rs6025)
Patients with cancer often suffer from a systemic activation of blood coagulation that can be attributed to the cancer itself or to its treatment. This excess of coagulation is reflected in increased risk of venous thrombosis, and it is widely accepted that the hemostatic system participates in neovascularization, tumor growth and metastatic spread, especially through the signaling of the tissue factor pathway. (TF) [28]. Activated protein C (APC) is an enzyme that is generated from protein C of the zymogen by the action of thrombin bound to thrombomodulin, which participates in the negative regulation of coagulation. The reduction of sensitivity to the anticoagulant function of APC, commonly referred to as resistance to APC, is an established risk factor for thrombosis in the general population [28].
APC resistance is mainly caused by Leiden factor (V) polymorphism (F5 rs6025), which leads to poor inactivation of FVa, as well as impaired APC-cofactor activity of FV in the inactivation of FVIIIa [28]. The presence of acquired resistance APC has been shown in several types of cancer, including breast cancer, which may increase the risk of venous thrombosis associated with cancer in patients with breast cancer [28].
ABCG2
ATP-binding cassette transport proteins (ABCs) comprise a large superfamily of membrane proteins that are present in all living organisms from bacteria, fungi, plants and animals [29]. It has been identified that overexpression of ABC multispecific transporters plays an important role in the resistance of tumors to chemotherapeutic drugs, which leads to insufficient concentrations of the drug within the cancer cell, resulting in a failure of the therapy [ 29].
The mechanism by which this process is carried out is due to the fact that the ABC transporters use the energy of ATP hydrolysis to expel their substrates from the cell, commonly against a concentration gradient, promoting a conformational change and facilitating the active efflux of a variety of anticancer substrates [30]. Because structurally unrelated drugs are affected by this type of resistance, it is called multidrug resistance (MDR) [29,30].
ABCG2 also known as breast cancer resistance protein is a transporter medium consisting of 655 amino acids and possessing only one cytosolic nucleotide binding domain (NBD) and one transmembrane domain (TMD) [29].
ABCG2 recognizes and transports numerous anticancer drugs, including conventional chemotherapeutic molecules and small targeted therapeutic molecules such as the cytostatic mitoxantrone and camptothecin derivatives topotecan and irinotecan [29]. It is also able to expel some photosensitizers such as feoforbide A and protoporphyrin IX suggesting that ABCG2 is a possible cause of cellular resistance to photodynamic therapy in cancer [29].
Kruppel-like factor 4 (KLF4)
The Kruppel-like factor 4 is a transcription factor that participates in the regulation of various cellular processes such as cell growth, proliferation and differentiation, as well as the self-renewal of stem cells and the maintenance of pluripotency [31].
KLF4 possesses a transactivation domain and a repression domain, which together determine the specificity of the transcriptional regulatory activity of KLF4 through interaction with other factors and modulation of DNA binding efficiency [31, 32]. The activity of KLF4 is negatively regulated by the phosphorylation of ERK1 and ERK2 in serine 132, which results in the induction of differentiation of embryonic stem cells [32]. It has been shown that the expression of KLF4 is a poor prognostic factor for early breast and skin cancer, which corroborates its oncogenic role, it has also been shown that some of Krüppel's factors, such as KLF4 and KLF15, can suppress the progression of ERα-dependent cancer [33].
The level of expression of KLF4 and ERα are positively correlated and patients who are sensitive to treatment with tamoxifen show a higher expression of KLF4, as well as a better disease-free survival after treatment. In vitro experiments have shown that KLF4 levels in tamoxifen-resistant cell lines are low, and that a higher expression of KLF4 leads to an increased sensitivity to tamoxifen [33]. It is proposed that KLF4 contributes to the sensitivity of tamoxifen in breast cancer through the modification of the phosphorylation of ERK and p38 signalling and highlights the importance of the interaction of the KLF4 / MAPK signal to regulate the tamoxifen resistance of the breast cancer [33].
Cancerous inhibitor of the protein phosphatase 2A (CIP2A)
CIP2A is an inhibitor of cellular protein phosphatase 2A (PP2A), which contributes to the immortalization and malignant transformation of human cells [34]. It has been determined that the levels of this protein are over-expressed in human breast cancer tissues, so its presence allows the diagnosis and prognosis of breast cancer, therefore it is useful as a diagnostic marker for this disease, in addition to being detectable in blood, which gives it greater value, as it is easily detectable [3, 4]. It has been found that tamoxifen-induced apoptosis in breast cancer is associated with the down-regulation of CIP2A and p-AKT, likewise the membranous expression of CIP2A is significantly higher in patients with tamoxifen resistance, however the mechanism by which resistance is generated is not yet fully glimpsed [35].
Membranous CIP2A is an indicator of resistance to tamoxifen in breast cancer with positive hormone receptors. It is considered that it may be useful to select patients with positive estrogen receptor at risk of developing resistance to tamoxifen [35].
p95HER2
T-DM1 is an antibody-drug complex used as a new tool for the treatment of breast cancer HER2 positive. It is a complex compound obtained by the conjugation of trastuzumab and the potent cytotoxic drug derived from Mayanism DM1, which is able to inhibit cell division and induce cell death [36].
The binding of T-DM1 to HER2-positive cells by said receptor allows the internalization of this complex by endocytosis and proceeds to its subsequent intraliposomal proteolytic degradation. However, its clinical activity is limited by inherent or acquired pharmacological resistance. [36]. The overexpression of p95HER2 is one of the molecular mechanisms of resistance to trastuzumab, since it has been shown that the expression levels of p95HER2 are sensitive but not specific in the determination of trastuzumab resistance.
The mechanism by which p95-HER2 exerts its action to provide resistance to T-DM1, is that this isoform of the HER2 receptor lacks a specific binding site to trastuzumab, so that the drug cannot exert its biological action [38].
Exostosin 1 (EXT1)
Exostoxin 1 is a type II transmembrane glycoprotein that is regulated by the endoplasmic reticulum (ER) and is involved in the biosynthesis of heparan sulphate (HS) on the cell surface [39]. It has been identified that in MCF7 / ADR cells (doxorubicin-resistant breast cancer cell line) the EXT1 glycoprotein is highly expressed, which is reflected in an increase in EMT expression (epithelial mesenchymal transition), an overexpression of P-gp (P-glycoprotein) and with-it greater resistance to chemotherapy [39].
This was checked by suppressing EXT1 mediated by siRNA in MCF7 / ADR cells, which eliminated heparan sulphate from the cell surface. In addition, it was observed that the epithelial mesenchymal tumour was repressed and the sensitization of these cells to the action of doxorubicin was increased [39]. These findings can be used as a new vision to define EXT1 as a new therapeutic target to counteract the chemoresistance of breast cancer in breast cancer patients with therapeutic resistance based on anthracyclines [39].
Cytochrome P450 2D6 (CYP2D6)
CYP2D6 is an enzyme that is part of cytochrome P450 that participates in the metabolism of tamoxifen, converting it to 4-hydroxy-Ndesmethyltamoxyphene (endoxifen), 4-hydroxy tamoxifen and N-demethyltamoxifen [40]. The relative antiestrogenic potencies of 4-hydroxy tamoxifen and endoxifen are comparable, but plasma concentrations of endoxifen are up to 10-fold higher in patients with functional CYP2D6. A problem that has been found is that there are people with different levels of functionality of this cytochrome, since it can present slow, intermediate and extensive metabolizers, which can compromise the levels of tamoxifen and its metabolites at the site of action and therefore generate a resistance to treatment [40].
Current and future focus of research on genes and proteins associated with chemotherapeutic resistance in breast cancer
At present, the analysis of genes and their expression are the specialty of pharmacogenomics, which has the potential to revolutionize patient care by opening the door to personalized medicine in the field of pharmacotherapy and increase the likelihood of providing the right patient with the correct dose of the correct medication, using the correct route at the right time, improving its effectiveness and minimizing resistance to treatment [41].
An important part to achieve this is the study of somatic and germline mutations, since these have a great impact on the prognosis of the disease and the response to therapy. Somatic mutations appear after an oncogenic process within the tumour tissue, whereas germline mutations are hereditary alterations found in the individual. These mutations can be used as risk biomarkers to optimize therapeutic decisions among the subpopulations of patients with breast cancer. Therefore, genetic information can be used both for the selection of effective therapy and to avoid treatments with an unacceptable risk of adverse reactions or resistance to chemotherapeutics [42].
Pharmacogenetics and pharmacogenomics determine the genetic differences to predict the safety, toxicity and efficacy of drugs between individuals and populations. Pharmacogenetics studies the variability in the response of the drug due to inheritance focusing on the genes involved in the metabolism of the drug, seeks to determine the genetic cause of unexpected drug responses by focusing on the genetic variation of pharmacokinetics (absorption, distribution, metabolism and excretion of the drug) as well as pharmacodynamics (receptors, channels and transporters of drug response) that are mechanisms that can participate in the development of resistance to treatment, while pharmacogenomics increases the understanding of the response to the drug by studying all the genes in the genome [42].
The analysis of the proteins involved in the resistance to chemotherapy is carried out through the use of proteomics, which is a complement for the investigation of the translation and modification of the genome and an effective tool for the comprehensive understanding of the expression of the genome [43].
However, pharmacogenomics and pharmacogenetics have not been sufficient to determine all the implications of genes and their expression in the development of resistance to treatment, which is why the post-genomic era has begun, which deals with the application of omics technologies to biomedical and pharmaceutical research. Compared to traditional research methods, the application of omics sciences allows the exploration of the genome, transcriptome and proteome more broadly with greater sensitivity which can provide solutions for the discovery and validation of therapeutic targets, toxicity, pharmacology, molecular diagnosis, prognosis, and personalized medical attention [43].
With the application of the omics sciences, new areas of interest for the study of drug action and resistance mechanisms have emerged. One of these areas is the pharmacometabolomic, which studies the changes in the metabolome of an individual patient caused by the administration of a drug. It also allows us to analyse the effects of the drug on certain metabolic pathways involved in the mechanisms of variation in response to treatment, including resistance mechanisms [43].
One more field of application that is in development is the interact omics, which is a discipline that deals with studying both the interactions and the consequences of these interactions between proteins and other molecules within a cell (proteins, nucleic acids, lipids, carbohydrates, drugs). Among the most important interactions to be studied are the protein-protein interactions, the protein-DNA interactions and the protein-DNA-drug interactions, which would help us to identify new mechanisms of resistance to chemotherapeutics [43].
Conclusion
Analysing the role of genes and proteins that compromise the effective response to a treatment against breast cancer is of vital importance, since this process of resistance generates that the patient does not respond to the treatment and his condition becomes more serious, leading to death.
The discovery of new genes and proteins associated with resistance to chemotherapeutics, as well as their mechanism of resistance, can become risk biomarkers and possible therapeutic targets to guarantee the effectiveness of the treatment and with it a better prognosis and greater probability of cure.
It is clear that the way to elucidate all the genes and proteins involved in resistance to the treatment of breast cancer is very long and complex, due to the multiple variants of treatments and types of breast cancer present, which exponentially increases the chances of finding new genes and proteins that participate in the intrinsic or acquired resistance to treatment against breast cancer.
It is important to identify that resistance to the different therapeutic agents used to treat breast cancer regardless of the type of it, not only is it attributed to a particular gene or protein, but to a whole network of interactions between genes and proteins. These networks have been studied by the interact omics, however we still do not have complete knowledge of how these interactions are carried out and the scope they have, not only in the treatment of breast cancer, but from the analysis of the causes that lead to the development of this disease.
Acknowledgements
Authors JGSB and EMA managed the analyses of the study through the PRODEP project DSA/103.5/16/10569.
Article Info
Article Type
Research ArticlePublication history
Received: Sat 04, Aug 2018Accepted: Sat 18, Aug 2018
Published: Wed 19, Sep 2018
Copyright
© 2023 Jonnathan Guadalupe . This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited. Hosting by Science Repository.DOI: 10.31487/j.COR.2018.02.006
Author Info
Corresponding Author
Jonnathan GuadalupeFaculty of Chemistry, Autonomous University of the State of Mexico (UAEMex), Toluca, Mexico
Figures & Tables
Table 1: Genes and proteins associated with chemotherapeutic resistance in different types of breast cancer
|
Gene/Protein resistance effectors |
Abbreviation |
Chemotherapeutics affected by resistance |
Type of cancer where resistance is presented |
|
Fosfoserin aminotransferase-1
|
PSAT-1 |
Tamoxifen |
Luminal A and B (RE+, RP+) |
|
SOX9 embryonic transcription factor
|
SOX9 |
Tamoxifen |
Luminal A and B (RE+, RP+) |
|
Initiation complex of the eIF4F translation |
eIF4F |
Tamoxifen |
Luminal A and B (RE+, RP+) |
|
Leptin |
LEP |
Tamoxifen |
Luminal A and B (RE+, RP+) |
|
Mucin-1 protein |
MUC-1 |
Bortezomib, trastuzumab, tamoxifen |
Luminal A and B (RE+, RP+), positive Her-2 |
|
Glutathione S-transferase P1 |
GSTP-1 |
Anthracyclines and taxanes such as: adriamycin, daunomycin, epirubicin, docetaxel, paclitaxel, cyclophosphamide and doxorubicin |
Breast carcinoma in general |
|
β-glucosidase |
GBA |
Paclitaxel |
Breast carcinoma in general |
|
ESR-1 Gene |
ESR-1 |
Aromatase inhibitors such as: letrozole, anastrozole, exemestane |
Luminal A and B (RE+, RP+) in postmenopausal women. |
|
miRNA |
miRNA221/222, miRNA-342-3p, miRNA-873 |
Tamoxifen |
Luminal A and B (RE+, RP+) |
|
Interleukin 33 |
IL-33 |
Tamoxifen |
Luminal A and B (RE+, RP+) |
|
Factor E2F8 |
E2F8 |
Cisplatin |
Breast carcinoma in general |
|
Gene/Protein resistance effectors |
Abbreviation |
Chemotherapeutics affected by resistance |
Type of cancer where resistance is presented |
|
Mucin 4 |
MUC4 |
Trastuzumab |
Positive Her-2 |
|
ATP binding cassette transport protein G2 |
ABCG2 |
Mitoxantrone and camptothecin, topotecan and irinotecan derivatives |
Metastatic breast cancer |
|
Survivin |
BIRC-5 |
Taxans, kinesin inhibitors and mTOR inhibitors |
Breast carcinoma in general |
|
Brachyury protein / SIRT1 gene |
T/SIRT1 |
Tamoxifen |
Luminal A and B (RE+, RP+) |
|
Membrane cell adhesion molecule |
MCAM CD146 |
Tamoxifen |
Luminal A and B (RE+, RP+) |
|
Cyclin E |
CCNE-1 |
Aromatase inhibitors such as: letrozole, anastrozole, exemestane |
Luminal A and B (RE+, RP+)in postmenopausal women. |
|
Nuclear factor enhancer of the kappa light chains of activated B cells |
NF-κB |
RTK inhibitors such as trastuzumab, bevacizumab or imatinib |
Breast cancer triple negative marker |
|
Krüppel-like factor 4 |
KLF4 |
Tamoxifen |
Luminal A and B (RE+, RP+) |
|
Cancerous protein phosphatase 2A inhibitor |
CIP2A |
Tamoxifen |
Luminal A and B (RE+, RP+) |
|
p95-HER2 |
p95-HER2 |
Trastuzumab T-DM1 |
Positive Her-2 |
|
Exostosin 1 |
EXT1 |
Doxorubicin |
Breast carcinoma in general |
|
Cytochrome P450 2D6 |
CYP2D6 |
Tamoxifen |
Luminal A and B (RE+, RP+) |
References
1. Zhang J, Kinoh H, Hespel L, Liu X, Quader S, Martin J et al. (2017) Effective treatment of drug resistant recurrent breast tumors harboring cancer stem-like cells by staurosporine/epirubicin co-loaded polymeric micelles. J Control Release. [Crossref]
2. Lal S, McCart Reed A, de Luca X, Simpson P (2017) Molecular signatures in breast cancer. Methods 131: 135-146. [Crossref]
3. Drăgănescu M, Carmocan C (2017) Hormone Therapy in Breast Cancer. Chirurgia 112: 413-417.
4. Wilhelmsson A, Roos M, Hagberg L, Wengström Y, Blomberg K (2017) Motivation to uphold physical activity in women with breast cancer during adjuvant chemotherapy treatment. Eur J Oncol Nurs 29: 17-22. [Crossref]
5. Seo H, Ku J, Lee H, Woo J, Cheon C, Kim M et al. (2017) SH003 reverses drug resistance by blocking signal transducer and activator of transcription 3 (STAT3) signalling in breast cancer cells. Bioscience Reports BSR20170125. [Crossref]
6. De Marchi T, Timmermans M, Sieuwerts A, Smid M, Look M, et al. (2017) Phosphoserine aminotransferase 1 is associated to poor outcome on tamoxifen therapy in recurrent breast cancer. Sci Rep 7: 2099. [Crossref]
7. Jeselsohn R, Cornwell M, Pun M, Buchwalter G, Nguyen M, et al. (2017) Embryonic transcription factor SOX9 drives breast cancer endocrine resistance. Proc Natl Acad Sci U S A 114: E4482-E4491. [Crossref]
8. Fagan D, Fettig L, Avdulov S, Beckwith H, Peterson M, et al. (2017) Acquired Tamoxifen Resistance in MCF-7 Breast Cancer Cells Requires Hyperactivation of eIF4F-Mediated Translation. Horm Cancer 8: 219-229. [Crossref]
9. Qian Y, Shi D, Qiu J, Zhu F, Qian J, et al. (2015) ObRb downregulation increases breast cancer cell sensitivity to tamoxifen. Tumour Biol 36: 6813-6821. [Crossref]
10. Farahmand L, Merikhian P, Jalili N, Darvishi B, Majidza K (2017) Significant role of MUC1 in development of resistance to currently existing anti-cancer therapeutic agents. Current Cancer Drug Targets 17.
11. Merikhian P, Ghadirian R, Farahmand L, Mansouri S, Majidzadeh-A K (2017) MUC1 induces tamoxifen resistance in estrogen receptor-positive breast cancer. Expert Rev Anticancer Ther 17: 607-613. [Crossref]
12. Yang S, Wang D, Li J, Xu H, Shen H, et al. (2017) Predictive role of GSTP1-containing exosomes in chemotherapy-resistant breast cancer. Gene 623: 5-14. [Crossref]
13. Zhou X, Huang Z, Yang H, Jiang Y, Wei W, et al. (2017) β-Glucosidase inhibition sensitizes breast cancer to chemotherapy. Biomed Pharmacother 91: 504-509. [Crossref]
14. Yates L, Desmedt C (2017) Translational Genomics: Practical Applications of the Genomic Revolution in Breast Cancer. Clin Cancer Res 23: 2630-2639. [Crossref]
15. Muluhngwi P, Klinge C (2017) Identification of miRNAs as biomarkers for acquired endocrine resistance in breast cancer. Mol Cell Endocrinol 456: 76-86. [Crossref]
16. Muluhngwi P, Klinge C (2015) Roles for miRNAs in endocrine resistance in breast cancer. Endocr Relat Cancer 22: R279-R300. [Crossref]
17. Shen H, Wang D, Li L, Yang S, Chen X, et al. (2017) MiR-222 promotes drug-resistance of breast cancer cells to adriamycin via modulation of PTEN/Akt/FOXO1 pathway. Gene 596: 110-118. [Crossref]
18. Hu H, Sun J, Wang C, Bu X, Liu X, et al. (2017) IL-33 facilitates endocrine resistance of breast cancer by inducing cancer stem cell properties. Biochem Biophys Res Commun. 485: 643-650. [Crossref]
19. Tian J, Lin Y, Yu J (2017) E2F8 confers cisplatin resistance to ER+ breast cancer cells via transcriptionally activating MASTL. Biomed Pharmacother 92: 919-926. [Crossref]
20. Li K, Ying M, Feng D, Du J, Chen S, et al. (2016) Brachyury promotes tamoxifen resistance in breast cancer by targeting SIRT1. Biomed Pharmacother 84: 28-33. [Crossref]
21. Liang Y, Zeng D, Xiao Y, Wu Y, Ouyang Y, et al. (2017) MCAM/CD146 promotes tamoxifen resistance in breast cancer cells through induction of epithelial–mesenchymal transition, decreased ERα expression and AKT activation. Cancer Lett 386: 65-76. [Crossref]
22. Doostan I, Karakas C, Kohansal M, Low K, Ellis M, et al. (2017) Cytoplasmic Cyclin E Mediates Resistance to Aromatase Inhibitors in Breast Cancer. Clin Cancer Res 23: 7288-7300. [Crossref]
23. Darvishi B, Farahmand L, Eslami-S Z, Majidzadeh-A K (2017) NF-κB as the main node of resistance to receptor tyrosine kinase inhibitors in triple-negative breast cancer. Tumour Biol 39: 101042831770691. [Crossref]
24. Mittal R, Jaiswal P, Goel A (2015) Survivin: A molecular biomarker in cancer. Indian J Med Res 141: 389. [Crossref]
25. Taglieri L, De Iuliis F, Giuffrida A, Giantulli S, Silvestri I, et al. (2017) Resistance to the mTOR inhibitor everolimus is reversed by the downregulation of survivin in breast cancer cells. Oncol Lett. [Crossref]
26. Duraisamy S, Ramasamy S, Kharbanda S, Kufe D (2006) Distinct evolution of the human carcinoma-associated transmembrane mucins, MUC1, MUC4 AND MUC16. Gene 373: 28-34. [Crossref]
27. Wimana Z, Gebhart G, Guiot T, Vanderlinden B, Larsimont D, et al. (2017) N-Acetylcysteine breaks resistance to trastuzumab caused by MUC4 overexpression in human HER2 positive BC-bearing nude mice monitored by 89Zr-Trastuzumab and 18F-FDG PET imaging. Oncotarget 8: 56185-56198. [Crossref]
28. Tinholt M, Sandset P, Mowinckel M, Garred, Sahlberg K, et al. (2016) Determinants of acquired activated protein C resistance and D-dimer in breast cancer. Thromb Res 145: 78-83. [Crossref]
29. Krapf M, Gallus J, Wiese M (2017) Synthesis and biological investigation of 2,4-substituted quinazolines as highly potent inhibitors of breast cancer resistance protein (ABCG2). Eur J Med Chem 139: 587-611. [Crossref]
30. Patel A, Li T, Anreddy N, Wang D, Sodani K, et al. (2017) Suppression of ABCG2 mediated MDR in vitro and in vivo by a novel inhibitor of ABCG2 drug transport. Pharmacol Res 121: 184-193. [Crossref]
31. Wang B, Zhao M, Cui N, Lin D, Zhang A, et al. (2015) Krüppel-like factor 4 induces apoptosis and inhibits tumorigenic progression in SK-BR-3 breast cancer cells. FEBS Open Bio 5: 147-154. [Crossref]
32. Ghaleb A, Yang V (2017) Krüppel-like factor 4 (KLF4): What we currently know. Gene 611: 27-37. [Crossref]
33. Jia Y, Zhou J, Luo X, Chen M, Chen Y, et al. (2017) KLF4 overcomes tamoxifen resistance by suppressing MAPK signaling pathway and predicts good prognosis in breast cancer. Cell Signal 42:165-175. [Crossref]
34. Xing M, Lu Y, Wang D, Zou X, Zhang S, et al. (2016) Clinical significance of sCIP2A levels in breast cancer. Eur Rev Med Pharmacol Sci 20: 82-91. [Crossref]
35. Baldacchino S, Wastall L, Saliba C, Hughes T, Scerri C, et al. (2017) CIP2A expression predicts recurrences of tamoxifen-treated breast cancer. Tumour Biol 39. [Crossref]
36. Martínez M, Pérez-Fidalgo J, Martín-Martorell P, Cejalvo J, Pons V, et al. (2016) Treatment of HER2 positive advanced breast cancer with T-DM1: A review of the literature. Crit Rev Oncol Hematol 97: 96-106. [Crossref]
37. Eliyatkin NO, Aktas S, Ozgur H, Ercetin P, Kupelioglu A (2016) The role of p95HER2 in trastuzumab resistance in breast cancer. J BUON 21: 382-389. [Crossref]
38. Sung M, Tan X, Lu B, Golas J, Hosselet C, et al. (2017) Caveolae-mediated endocytosis as a novel mechanism of resistance to trastuzumab emtansine (T-DM1). Mol Cancer Ther 17: 243-253. [Crossref]
39. Manandhar S, Kim C, Lee S, Hyun Kang S, Basnet N, et al. (2017) Exostosin 1 regulates cancer cell stemness in doxorubicin-resistant breast cancer cells. Oncotarget 8: 70521-70537. [Crossref]
40. Clarke R, Tyson J, Dixon J (2015) Endocrine resistance in breast cancer – an overview and update. Mol Cell Endocrinol 418: 220-234. [Crossref]
41. Khelifi M, Tarczy-Hornoch P, Devine EB, Pratt W (2017) Design Recommendations for Pharmacogenomics Clinical Decision Support Systems. AMIA Jt Summits Transl Sci Proc 2017: 237-246. [Crossref]
42. López-Cortés A, Guerrero S, Redal M, Alvarado A, Quiñones L (2017) State of Art of Cancer Pharmacogenomics in Latin American Populations. Int J Mol Sci 18: 639. [Crossref]
43. Yan SK, Liu RH, Jin HZ, Liu XR, Ye J, et al. (2015) "Omics" in pharmaceutical research: overview, applications, challenges, and future perspectives. Chin J Nat Med 13: 3-21. [Crossref]