Electrophysiological and Morphological Studies of SOD1 Transgenic Mice: An Animal Model of ALS
A B S T R A C T
Amyotrophic lateral sclerosis (ALS) is a disease where upper and lower motor neurons die, and it is often associated with mutations of superoxide dismutase 1 (SOD1). We have used mouse models to compare physiologic and morphologic characteristics of cervical motor neurons in wild-type and mutant animals. Slices of the cervical spinal cord were prepared from old wild-type and mutant G93A and G85R mice, and intracellular recordings of membrane potential, resistance and responses to application of excitatory neurotransmitters were studied. Some motor neurons were injected with Lucifer Yellow for morphological analysis. There were no significant differences between membrane potential in the SOD1 mutants and aged wild-type mice, but membrane resistance was somewhat higher in the mutant motor neurons. Dendrites of the mutant motor neurons were not responsive to ionophoretic application of excitatory amino acids, although the cell body responded strongly. In Lucifer-filled cells, the dendrites were found to disappear. Mutant motor neurons were sometimes spontaneously active. Responses of mutant motor neurons to perfused glutamate with varying calcium concentrations in the Ringer’s solution were different from those of the wild-type cells.
Keywords
Lucifer yellow CH, ALS, membrane potential, membrane resistance, dendrites, glutamic-acid subtypes
Introduction
In 1869, French neurologist, Jean-Martin Charcot discovered a disease in humans characterized by abnormalities and loss of skeletal muscle mass and named it amyotrophic-related lateral sclerosis (ALS) [1]. Most ALS is sporadic, but 5 to 10% is familial. Some 20 different mutations have been identified in familial ALS [2]. Mutations of superoxide dismutase 1 (SOD1) have been seen in about 20% of familial cases and 3% of sporadic cases [3, 4]. Mouse models of these mutations have been developed; the best studied being SOD1-G93A [5]. In the SOD1 mutant mouse, the muscle force begins to decline around about 150 days after birth [6].
Biochemical observations of the SOD1 mutant mice show mitochondrial dysfunction, SOD1 aggregation, motor neuron cell death and paralysis [7]. There is a striking increase in oxidative stress, as in common among other neurodegenerative diseases [8]. A major role of mitochondria is calcium regulation, and this is clearly disrupted in ALS. Reactive oxygen species (ROS) are generated by the intracellular influx of a large quantity of calcium going through both voltage and agonist activated calcium channels, and these processes play a major role in motor neuron cell death. Homeostasis of the concentration of intracellular calcium is lost [9]. There is also a fragmentation of the Golgi apparatus, which further disrupts cell function [10].
Glutamate is the major excitatory neurotransmitter in the central nervous system. There are several different glutamate receptors (AMPA, kainic acid and NMDA), but the AMPA receptor is the one responsible for usual synaptic transmission. This receptor usually consists of four subunits, GluA1, GluA2 GluA3 and GluA4), and the open ion channel is not permeable to calcium [11]. However, without RNA editing of GluA2, the AMPA receptor is permeable to calcium. Kwak and Kawahara (2005) have shown both in mice and humans that ALS is associated with abnormal editing of GluA2 and have provided support for the hypothesis that the motor neuronal cell death is due to elevation in calcium concentration [12, 13]. We have performed electrophysiologic and morphologic studies comparing wild-type cervical motor neurons to those of SOD1 mutant mice in order to investigate changes occurring in structure, membrane biophysical properties and responses to excitatory amino acid neurotransmitters during the period in which motor neurons are showing injury.
Methods and Materials
We studied motor neurons in the cervical spinal cord of 55 G93A mutant mice, 20 G85R mutant mice and 22 wild-type mice. We used methods described in previous studies on rat spinal cord and those described for mouse in the accompanying paper [14, 15]. The vertebra surrounding the spinal column of mice is not as hard as in the rat and can be removed without damaging the spinal cord. It is also easier to remove remnants of the surrounding dura mater in comparison with an albino rat. The size of a mouse is smaller than a rat, and also the skeleton is thinner. We perfused cold Ringer’s solution over the cord during dissection, which significantly reduced the rate of metabolism. Only motor neurons were studied that had a membrane potential of about -60 mV and for which stable recordings were maintained for a period of at least 20 min.
As already reported, we removed an approximate 7 mm length of the cervical spinal cord around the biggest part (C-5), and we prepared slices of about 450-microns [14, 15]. The slices were preincubated in a modified Krebs-Ringer solution for about one hour. The composition of the modified Krebs-Ringer solution was 212.5 mM sucrose, 3.5 mM KCl, 2.4 mM CaCl, 1.3 mM MgSO4, 26 mM NaHCO3, 1.2 mM KH2PO4 and 10 mM glucose saturated with 95% O2/5% CO2 [16]. During the recording, the composition of the Krebs-Ringer solution was 125 mM NaCl, 3.5 mM KCl, 2.4 mM CaCl2, 1.3 mM MgSO4, 26 mM NaHCO3, 1.2 mM KH2PO4 saturated with 95% O2/5% CO2. Recordings were carried out in a submerged recording chamber perfused by normal Krebs-Ringer solution circulating at about 5 ml/min at 32°C. Electrical membrane properties of motor neurons were measured in the slice preparations by using conventional glass microelectrodes connected to a DC amplifier (Neuro Data) with a bridge circuit [14]. The glass micropipettes were made from borosilicate capillary tubing was filled with 3 M potassium acetate and having a resistance of about 150 MΩ.
Chemicals were added to the perfused Ringer’s solution or were placed in a glass micropipette and applied into the dendrite trees of the motor neuron ionophoretically. The chemicals used in these studies were excitatory amino acid agonists. Glutamate, AMPA and kainic acid (KA) were prepared as a 10 mM solution in 0.15 M NaCl in pH 7.5. For morphological analysis, microelectrodes were filled with Lucifer yellow CH (Sigma, 10% in distilled water) and were injected into motor neurons in the slices by passing current (2 nA negative current pulses of 250 ms at 2 Hz for 1,5-2 min). All studies of both SOD1 mutants and wild-type controls were done on mice at about post-natal day (PD) 125 to PD250. At these ages, both of the SOD1 mutant mice were showing a significant decrease in muscle force [6]. To the degree possible we compared mutant and wild-type responses in animals as close as possible to the same ages. We observed no significant differences in results obtained from the G93A as compared to the G85R mutant motor neurons, and therefore for some results, we pooled the mutant data for comparison with the wild-type.
Results
Figure 1 shows membrane potential and membrane resistance from motor neurons recorded from each mutant animal and their wild-types. Input resistance was significantly higher in the motor neurons from both mutant mice, although there was no significant difference between mutant and wild-type membrane potential. Figure 2 shows the structure of cervical motor neurons from wild-type mice at ages between PD150 to PD215 and of G93A mutant mice at ages PD143 to PD306. Wild-type motor neurons show extensive dendritic trees, but for the mutant animals, there is a large loss of dendrites with age. At PD306 there are effectively no dendrites left. Figure 3 shows responses to ionophoretic applications of excitatory amino acid neurotransmitters onto motor neurons from wild-type and G93A animals. The ionophoretic electrode, which was filled with neurotransmitter, was placed about 300 μm away from the cell body, which is the region where dendrites should be located, as confirmed by intracellular injection of Lucifer yellow [17]. As expected, wild-type motor neurons were depolarized by the amino acid applications, leading to generation of action potentials. In contrast with an even significantly larger current, the mutant motor neurons showed some depolarization but failed to generate any action potentials.
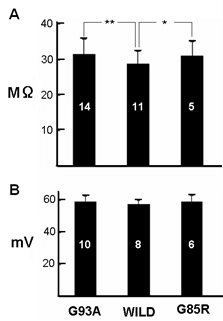
Figure 1: Histogram of A) membrane potential and B) input resistance from motor neurons from G93A, G85R and wild-type mice. Numbers in the columns indicate the number of neurons tested at around PD250. For input resistance; p < 0.05 for *, **.

Figure 2: Morphological observation of wild-type (PD125-215) and G93A (PD149-306) motor neurons by Lucifer-Yellow injection.

Figure 3: Intracellular recordings showing effects of ionophoretic application of the glutamate agonists, AMPA (A) and KA (K) and glutamate (G) at a site about 300 µm away from the cell body of a single motor neuron from a & b) a wild-type mouse at PD125 and c) a G93A mutant mouse at PD152. The applied current to release the drugs is indicated below the responses. The reaction to ionophoresis into the area where the dendrites should be located in the G93A motor neuron was very much weaker. Note that even with a much larger applied current the mutant motor neuron is not depolarized enough to generate an action potential.
Figure 4 shows a similar experiment but with bath perfusion of the excitatory amino acids onto cervical slices from wild-type and G93A mice. With bath perfusion, the mutant motor neurons are very responsive to AMPA and KA even at a lower concentration. These results show that the cell body still has electrical excitability and has excitatory amino acid receptors, even though the dendritic function of SOD1 mutant mouse has been very much reduced. Figure 5 shows the results of the application of intracellular current in a motor neuron from a wild-type and mutant animal. Intracellular recordings are from the cell body, not dendrites. The amount of current necessity to evoke an action potential is much less in the mutant motor neuron than the wild-type animal. Figure 6 shows an intracellular recording from a cervical motor neuron from a G93A mouse at age PD195. This motor neuron shows spontaneous pacemaker-like activity which is either i) increased or decreased by ii) depolarizing or hyperpolarizing current, respectively. Spontaneous activity was only seen in a few motor neurons but is particularly interesting because in early ALS spontaneous twitching of muscles is common, reflecting the repetitive discharge of a single motor neuron with contraction of all of the muscle fibers of that motor unit.
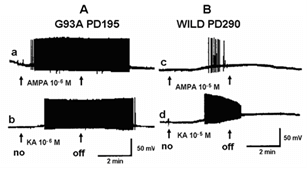
Figure 4: Intracellular recordings showing effects of bath perfusion of the glutamate agonists AMPA and KA on a motor neuron of a & b) a G93A animal (PD195) and c & d) a wild-type animal at PD290. Bath perfusion of a) AMPA (10-6 M). b) KA (10-6 M). c) AMPA (10-5 M). d) KA (10-5 M). The arrows show the on and off of the perfusion of the drug. Note that the concentrations applied to the mutant motor neuron is ten-times lower than that to the wild-type.
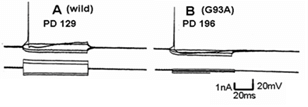
Figure 5: The difference in threshold value for excitation in response to current injection in the cell. A) Wild-type. B) SOD1 mutant mouse. The upper trace records potential while the lower trace is applied current.
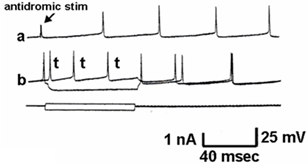
Figure 6: Typical electrical recording of a motor neuron from a G93A PD195 mouse showing spontaneous firing. a) The arrow shows the artifact of an antidromic stimulation that failed to generate a full action potential. The others are spontaneous discharges. b) This shows inhibition of the spontaneous discharge by hyperpolarizing current and an increase in the action potentials with the depolarization.
Figure 7 shows the effects of perfusing cervical slices with Ringer’s solution containing different concentrations of calcium on the AMPA responses of wild-type and G93A motor neurons. The wild-type motor neuron response did not change dramatically in solutions with different calcium concentrations. However, the response of the mutant motor neuron became very unstable with higher calcium concentrations.
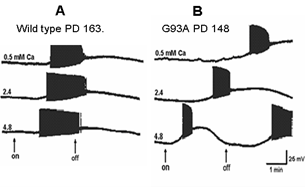
Figure 7: Influence of different calcium concentrations in the Ringer’s solution on responses to perfused AMPA (10-5M) in A) a wild-type motor neuron from an animal at PD163 and B) a G93A animal at PD148. The concentration of calcium was varied between 0.5, 2.4 and 4.8 mM. The arrows show on and off of the AMPA perfusion.
Discussion
The most striking feature we find when comparing cervical motor neurons of wild-type and SOD1 mutant mice are the selective loss of the dendritic tree while the cell body remains active, even generating pacemaker-like action potentials. The loss of dendrites is shown dramatically in the Lucifer-Yellow filled cells. Fogarty et al. (2016) have previously reported the loss of synaptic spines and dendrites in SOD1 mutant mice, but from cortical pyramidal neurons, not motor neurons [18]. The changes in electrical properties of the motor neurons are consistent with this observation. Membrane potential, always measured in the cell body, was not significantly different between wild-type and mutant motor neurons. However, membrane resistance was significantly greater in the mutants. This is what one would expect with loss of dendrites. It is also of interest that even when there has been a significant loss of dendrites, the motor neuron cell body remains electrically excitable and responsive to excitatory amino acid receptors. If anything, the cell bodies are unusually excitable, even to the point of generating spontaneous pacemaker discharges. While spontaneous pacemaker activity is common in invertebrate neurons, it has not often been observed in mammalian neurons. However, we have previously found that neurons in the medial vestibular nucleus are endogenous pacemakers and spontaneously active neurons have been recorded in spinal cord slices [19, 20]. In the case of a spontaneous motor neuron, one would expect to see contractions of the muscle fibers innervated by that motor neuron. Repetitive twitching of motor units is common in ALS patients [21].
As shown in (Figure 5), the threshold for action potential generation in the mutant motor neuron is low in comparison with the wild-type. Since the membrane potential in the mutant motor neuron is almost the same as in the wild-type, this may be a result of the difference in input resistance. Calcium, both extracellular and intracellular, plays a very important role in regulation of normal neuronal function and is a factor in neuronal damage and death [22]. This is why we studied the effects of varying extracellular calcium concentration on the responses to AMPA on wild-type and mutant mice. The response of AMPA with increasing calcium concentration in the perfused Ringer’s in the wild-type cell was a gradually increased response, but an abnormal excitation was seen in the mutant cell. We hypothesize that the reason for this excessive excitation relates to intracellular calcium regulation. The mitochondria have a close relation to the endoplasmic reticulum and together, they control the concentration of intracellular calcium [23]. The rhythmical change of depolarization and hyperpolarization of the membrane potential is probably the absorption and release of the calcium between the endoplasmic reticulum and mitochondria [23]. However, further study of these effects is needed.
Conclusion
We have compared the morphologic and electrophysiologic responses of cervical motor neurons of wild-type and two mouse mutants that are good models of the human disease, ALS. The motor neurons of SOD1 mutant mice show a progressive loss of dendrites but the cell body remains hyperexcitable, generating increased sensitivity to excitatory neurotransmitters applied to the cell body and even generating spontaneous pacemaker discharges.
Ethical Approval
This work was approved by the Animal Research Review Board of the University at Albany.
Article Info
Article Type
Research ArticlePublication history
Received: Mon 16, Aug 2021Accepted: Tue 31, Aug 2021
Published: Sat 02, Oct 2021
Copyright
© 2023 David O. Carpenter. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited. Hosting by Science Repository.DOI: 10.31487/j.JBN.2021.01.03
Figures & Tables
References
1. Ma YL, Li ZP (2019)
[Jean-Martin Charcot, discovery and nomenclature of amyotrophic lateral
sclerosis]. Zhonghua Yi Shi Za Zhi 49: 14-18. [Crossref]
2. Corcia P, Couratier
P, Blasco H, Andres CR, Beltran S et al. (2017) Genetics of amyotrophic lateral
sclerosis. Rev Neurol (Paris) 173: 254-262. [Crossref]
3. Kaur SJ, McKeown
SR, Rashid S (2016) Mutant SOD1 mediated pathogenesis of Amyotrophic Lateral
Sclerosis. Gene 577: 109-118. [Crossref]
4. Rosen DR, Siddique
T, Patterson D, Figlewicz DA, Sapp P et al. (1993) Mutations in Cu/Zn
superoxide dismutase gene are associated with familial amyotrophic lateral
sclerosis. Nature 362: 59-62. [Crossref]
5. Lutz C (2018) Mouse
models of ALS: Past, present and future. Brain Res 1693: 1-10. [Crossref]
6. Kong J, Xu Z (1998)
Massive mitochondrial degeneration in motor neurons triggers the onset of
amyotrophic lateral sclerosis in mice expressing a mutant SOD1. J Neurosci
18: 3241-3250. [Crossref]
7. Kim G, Gautier O,
Tassoni Tsuchida E, Ma XR, Gitler AD (2020) ALS Genetics: Gains, Losses, and
Implications for Future Therapies. Neuron 108: 822-842. [Crossref]
8. Islam MT (2017)
Oxidative stress and mitochondrial dysfunction-linked neurodegenerative
disorders. Neurol Res 39: 73-82. [Crossref]
9. Kawamata H,
Manfredi G (2010) Mitochondrial dysfunction and intracellular calcium
dysregulation in ALS. Mech Ageing Dev 131: 517-526. [Crossref]
10. Mourelatos Z, Adler
H, Hirano A, Donnenfeld H, Gonatas JO et al. (1990) Fragmentation of the Golgi
apparatus of motor neurons in amyotrophic lateral sclerosis revealed by
organelle-specific antibodies. Proc Natl Acad Sci U S A 87: 4393-4395. [Crossref]
11. Atoji Y, Sarkar S
(2019) Localization of AMPA, kainate, and NMDA receptor mRNAs in the pigeon
cerebellum. J Chem Neuroanat 98: 71-79. [Crossref]
12. Kwak S, Kawahara Y
(2005) Deficient RNA editing of GluR2 and neuronal death in amyotropic lateral
sclerosis. J Mol Med (Berl) 83: 110-120. [Crossref]
13. Kwak S, Hideyama T,
Yamashita T, Aizawa H (2010) AMPA receptor-mediated neuronal death in sporadic
ALS. Neuropathology 30: 182-188. [Crossref]
14. Hori N, Tan Y,
Strominger NL, Carpenter DO (2001) Intracellular activity of rat spinal cord
motoneurons in slices. J Neurosci Methods 112: 185-191. [Crossref]
15. Hori N, Xu Z,
Akaike N, Tan Y, Carpenter DO (2021) Physiological and pharmacological studies
on cervical motor neurons in slices prepared from neonatal and aged mice.
16. Aghajanian GK,
Rasmussen K (1989) Intracellular studies in the facial nucleus illustrating a
simple new method for obtaining viable motoneurons in adult rat brain slices. Synapse
3: 331-338. [Crossref]
17. Hori N, Tan Y, King
M, Strominger NL, Carpenter DO (2002) Differential actions and excitotoxicity
of glutamate agonists on motoneurons in adult mouse cervical spinal cord slices.
Brain Res 958: 434-438. [Crossref]
18. Fogarty MJ, Mu EWH,
Noakes PG, Lavidis NA, Bellingham MC (2016) Marked changes in dendritic
structure and spine density precede significant neuronal death in vulnerable
cortical pyramidal neuron populations in the SOD1(G93A) mouse model of
amyotrophic lateral sclerosis. Acta Neuropathol Commun 4: 77. [Crossref]
19. Lin Y, Carpenter DO
(1993) Medial vestibular neurons are endogenous pacemakers whose discharge is
modulated by neurotransmitters. Cell Mol Neurobiol 13: 601-613. [Crossref]
20. Carp JS, Tennissen
AM, Liebschutz JE, Chen XY, Wolpaw JR (2010) External urethral sphincter
motoneuron properties in adult female rats studied in vitro. J Neurophysiol 104: 1286-1300. [Crossref]
21. Ahmed A, Wicklund
MP (2011) Amyotrophic lateral sclerosis: what role does environment play? Neurol
Clin 29: 689-711. [Crossref]
22. Hori N, Carpenter DO (1994) Functional and morphological changes induced by transient in vivo ischemia. Exp Neurol 129: 279-289. [Crossref]
23. Ishii K, Hirose K, Iino M (2006) Ca2+ shuttling between endoplasmic reticulum and mitochondria underlying Ca2+ oscillations. EMBO Rep 7: 390-396. [Crossref]