ApoL6 Induces Dichotomous Cell Death Phenotype Involving Both Apoptosis and Necroptosis in Cancer Cells
A B S T R A C T
Apolipoprotein L6 (ApoL6) is a pro-death, BH3-only and phospholipid-binding member of the Bcl-2 family. Previously, we showed that ectopic expression of ApoL6 induces mitochondria-mediated apoptosis. In this study, we hypothesized that ApoL6 plays a dichotomous role in regulated cell death (RCD) pathways that involves both apoptosis and necroptosis. Necroptosis, or programmed necrosis, can occur simultaneously or alternatively to apoptosis as a caspase-independent pathway, and is characterized by the formation of intracellular necrosomes and plasma membrane pores. The pseudokinase mixed lineage kinase domain-like protein (MLKL) interacts with the necrosome to become phosphorylated and activated. Phosphorylated (p-) MLKL molecules migrate to the plasma membrane to form pores, disrupt membrane integrity, and selectively release intracellular components. The proinflammatory cytokine IL-1β is amongst a number of molecules released in this process. To investigate the role of ApoL6 in RCD, ApoL6 was overexpressed via a Tet-Off gene inducible system in a p53 null colorectal cancer cell line DLD-1. Necroptotic cell death was marked by an increased level of intracellular and plasma membrane p-MLKL and extracellular release of IL-1β along with the disruption of the plasma membrane. We found that in the presence of apoptotic inhibitors, levels of necroptotic cell death increased, indicating an enhanced push away from apoptosis. We also showed that ApoL6 preferentially binds with phosphatidylinositol phosphate species (PIPs), which include PI (4,5) P2, PI4P and PI5P. Thus, we expand on the role of ApoL6 in RCD to induce a mixed cell-death phenotype that includes both apoptosis and necroptosis.
Keywords
ApoL6, apoptosis, necroptosis, MLKL, IL-1β
Introduction
Regulated cell death (RCD) is an essential part of life for all multi-cellular organisms, playing a key role in a diverse array of processes throughout the body. RCD must proceed in an organized and orchestrated manner in order to ensure homeostasis and the removal of non-functioning cells while maintaining healthy cells [1-5]. This is different than necrosis wherein cell death is accidental, chaotic, and unregulated. Dysregulated RCD can result in several life-threatening diseases including cancer and inflammatory disorders [6-7]. When programmed cell death was initially discovered, it was solely in the context of apoptosis, wherein cells die through a caspase-dependent manner [8, 9]. Apoptosis is characterized by DNA fragmentation, chromatin condensation, membrane blebbing and cell-shrinkage [10].
Since the first mention of apoptosis, the study of regulated cell death has expanded to include the discovery of multiple possible pathways by which a cell can proceed to die in a non-apoptotic manner. These pathways include autophagy, pyroptosis, necroptosis and ferroptosis [11-21].
Necroptosis is an RCD pathway in response to cellular stress involving inhibition of apoptosis, particularly a caspase-8 independent means of activation. Necroptosis can be activated in response to tumor necrosis factor (TNF) as well as other factors such as TRAIL and Fas complemented by caspase-8 inhibition [22, 23]. The execution of necroptosis is facilitated by the critical regulators, receptor interacting protein kinase 1,3 (RIPK1, RIPK3) and the pseudokinase mixed lineage kinase domain-like protein (MLKL) [24-29]. Activated RIPK1 forms a complex with activated RIPK3 known as the necrosome which functions as an amyloid signaling complex to recruit and phosphorylate MLKL via interactions with activated RIPK3 [30]. Activated and phosphorylated MLKL (p-MLKL) oligomerizes and develops pores on the plasma membrane in order to permeabilize it and allow for necroptosis to proceed. It has been shown that p-MLKL migrates to the plasma membrane where it encounters and binds phosphatidylinositol phosphate species (PIPs) [31]. Binding is facilitated by charge-charge interactions between the negatively charged PIPs on the membrane and the positively charged four-helical bundle domain on MLKL. MLKL preferentially binds to PI (4,5) P2, PI4P and P5P in the plasma membrane [31-32]. This binding promotes oligomerization of p-MLKL and permeabilization of the plasma membrane allowing for the membrane rupture characteristic of necroptosis [32].
Necroptosis is distinguished from apoptosis by a number of biochemical and molecular characteristics, as well as in its ability to promote the selective release of intracellular components and signaling molecules to initiate an immune or inflammatory response [33, 34]. Upon membrane permeabilization by p-MLKL binding membrane lipids followed by oligomerization, pores are created. The opening of cells allows for the release of intracellular components including pro-inflammatory molecules such as the cytokine interleukin-1β (IL-1β), along with other members of the IL-1 family and damage associated molecular patterns (DAMPS) [35]. IL-1β is a key regulatory cytokine of the inflammatory and adaptive immune response, synthesized as an inactive form, pro-IL-1β- by affected cells [36]. Pro-IL-1β is cleaved by caspase-1 and rapidly converted to the active form and secreted through membrane pores. RIPK1, the crucial regulator of necroptosis has been documented to also be directly involved in the processing of pro-IL-1β [37-39]. In the context of necroptosis, IL-1β is released in the very end stages of this cell death pathway to function as a signaling molecule to other cells and enhance the immune and inflammatory response.
Apolipoprotein L6 (ApoL6) is a pro-death, BH3-only and lipid-binding member of the Bcl-2 family, a group of proteins that are critical regulators of cell death and homeostasis [40-43]. Previously, we also established a role for ApoL6 in promoting apoptosis and blocking beclin-1 dependent autophagy in atherosclerotic cells [41]. In atherosclerotic lesions, expression of ApoL6 is inducible by IFNγ and was found to colocalize with activated caspase-3 in smooth muscle cells. ApoL6 carries out its pro-apoptotic function via binding to the anti-apoptotic Bcl-xL and promoting ROS generation. Recently, ApoL6 was suggested to have a role in obesity and related cancers as it was shown to be the target of miR10b-5p where upregulation of ApoL6 was found to promote adipogenesis [44].
Importantly, we have shown previously that ectopic expression of ApoL6 induces mitochondria-mediated apoptosis via the release of cytochrome c along with SMAC/DIABLO from the mitochondria and the activation of caspases 8 and 9 [40]. This release of SMAC from the mitochondria prompted the investigation of a role for ApoL6 in necroptosis examined in the present study. Moreover, we postulated that ApoL6 could have a role in the connection of PIP trafficking and cell death. The recurring idea was also tested here to determine whether ApoL6 is involved in lipid binding and trafficking. In this study we primarily used the DLD1.ApoL6, p53-null colorectal cancer cell line that is gene inducible by the Tet-off system. P53-null and therefore p53-independence is an important characteristic that renders DLD-1 ideal for cell-death study by the effect of a single gene [45]. We demonstrated that ApoL6 induces a mixed cell death phenotype that includes apoptosis and necroptosis. We provided a link to necroptosis via the above-mentioned characteristics: cellular morphology, p-MLKL upregulation and migration to the plasma membrane, 1L-1β release, and PIP-binding.
Methods and Materials
I Cell Line, Medium and Drug Treatment
DLD-1.ApoL6 cells, a previously established, Tet-off inducible, p53-null colorectal cancer cell line, were cultured in D.20 “normal” medium made up of McCoy’s 5A supplemented with 10% FBS, 1X antibacterial antimycotic solution, and 20 ng/ml Doxycycline (Dox; D.20). Cells were grown to 60% confluence, rinsed with PBS and the media exchanged for a similar media containing lower concentrations of doxycycline, ranging from 0-20 ng/ml. Under these conditions, ApoL6 is overexpressed and cells will proceed to die [40, 41]. We determined that DLD-1.ApoL6 has an I.C.50 at a doxycycline concentration of 0.5 ng/mL (D.0.5) to achieve 50 % cell death in 6 hours. This concentration was used in subsequent experiments as the “induction” medium. Cells were treated with a combination of two drugs in order to manipulate the pathway by which cell death proceeds, both of which were subject to a 1:1000 dilution upon addition to the D.0.5 media. Z-VAD (10 µM; R&D Systems) is a strong caspase inhibitor and therefor an inhibitor of apoptosis. Cells subjected to this treatment were redirected towards the pathway of necroptotic cell death.
The addition of BV-6 (12.5 µM; Calbiochem), a SMAC mimetic and an antagonist of cIAP and XIAP, has a combined effect with Z-VAD to enhance the push away from apoptosis and shift cell death to proceed through the necroptotic pathway. Previously, we used other cell lines including an adenovirus ApoL6 overexpression system in the presence of caspase inhibitors that showed similar results to those presented here and prompted the examination of ApoL6 as an inducer of necroptosis [40].
II Scanning Electron Microscopic Imaging
Indicated cells were processed by primary fixation in 0.1 M sodium cacodylate buffer containing 3% formaldehyde and 2% glutaraldehyde followed by cacodylate buffer washes. Samples were then incubated in 2% lysine for 1 hour at room temperature, washed, then post-fixed in 1% osmium tetroxide for 1 hour [46]. Samples were then washed, dehydrated in a graded series of ethanol, further dehydrated in hexamethyldisilazane (HMDS), and air-dried. The coverslips were then mounted on stubs, sputter-coated with carbon or gold-palladium (Au-Pd) and examined in a Zeiss Sigma 300 field emission scanning electron microscopy (FE-SEM).
III Protein Extraction
To examine intracellular protein concentrations, DLD1.ApoL6 was cultured in a P100 culture dish until reaching ~60% surface area coverage. The cells were rinsed with PBS then treated for 6 hours with either D.20 normal media, D.0.5 induction media or D.0.5 with the addition of Z-VAD and BV-6. After the treatment was completed, the plate was put on ice and washed with cold PBS. The cells were then gently scraped off the plate using cold PBS and a rubber policeman and pipetted into a microfuge tube. The cell suspension was centrifuged at 2000 rpm at 4°C. The cell pellet was resuspended in RIPA [Tris-HCl, NaCl, sodium deoxycholate, SDS, Triton X-100] along with protease/phosphatase inhibitor cocktail. The resulting suspension was briefly sonicated and kept on ice followed by centrifugation. A Micro BCA Protein Assay Kit (Thermo Scientific) with standard protein solutions was used to assess protein concentrations. Concentrations were subsequently measured on a Nanodrop instrument.
IV SDS-PAGE and Western Blotting Analysis
Sixty micrograms of proteins were separated in each lane using 4-15% SDS–PAGE (Bio-Rad Mini-PROTEAN) and transferred to PVDF membranes via a Trans-Blot Turbo Transfer System (Bio-Rad). The membranes were blocked in 5% milk or BSA in 1x TBST, incubated with primary antibodies, p-MLKL (rabbit polyclonal 1:1,000, Cellsignal, mAb #91689) MLKL (rabbit polyclonal 1:1,000, Abcam, ab194699), or anti- β -actin (Sigma, A1978), and then probed with corresponding goat anti-rabbit or anti-mouse HP-labeled secondary antibodies (Bio-Rad). After washing with 1X TBST, ECL was added directly to the membrane and imaged (Bio-Rad Imager).
V ELISA Analysis of Extracellular IL-1β
In order to determine extracellular release of IL-1β, we utilized a 96-well IL-1β (Human) ELISA Kit (Avida Systems Biology). DLD1.ApoL6 were regrown in 6-well plates and put through the same drug treatments as previously described. Samples included a D.20, D.0.5 and D.0.5 + Z-VAD +BV-6 (1:1,000). After 6 hours, the media from all samples was collected and spun down at 2000 rpm for 5 minutes, collecting the supernatant immediately after centrifugation. 100 µL of samples, along with 8 standards ranging in concentration from 0 pg/mL-250 pg/mL of IL-1β, were added to the 96-well plate precoated with anti- IL-1β antibody and incubated at 37°C for 90 minutes. The media was replaced with 100 µL biotinylated 1X-IL1β and incubated at 37°C for 60 minutes. Wells were washed with buffer and 100 µL 1X Avidin Biotin Peroxidase was added and incubated for an additional 30 minutes at 37°C. 90 µL TMB colour developing agent was added after washing and incubated for 25 minutes at 37°C. Finally, 100 µL TMB stop solution stopped the reaction and absorbance was measured at 450 nm.
VI Immunofluorescence Microscopy of DLD-1.ApoL6
To study the subcellular localization of p-MLKL under apoptotic and necroptotic conditions, we preformed immunofluorescence microscopy of DLD-1.ApoL6. Cells were cultured in D.20 on glass coverslips pre-coated with poly-L-lysine in a 6-well plate. Medium was exchanged for either D.0.5 or D.0.5+ Z-VAD+BV-6 and incubated at 37°C for 6 hours. Coverslips were rinsed with PBS and treated with Cell Mask Deep Red (1:1,000), a plasma membrane specific marker. Cells were then fixed in 4% paraformaldehyde and rinsed with PBS. Blocking solution of 3% BSA was applied for 1 hour at room temperature then treated with primary antibody (anti-p-MLKL rabbit monoclonal 1:100) for 3 hours at room temperature. After rinsing, coverslips were treated with secondary antibody (Alexa 488 anti-rabbit 1:500 in 3% BSA) at room temperature for 2 hours in the dark. Coverslips were rinsed, mounted on slides and imaged on an EVOS fluorescence microscope using a 20x objective with a GFP and CY-5 light filter.
VII ApoL6 Binding Lipid Analysis
To determine ApoL6 phospholipid binding partners, we used an in vitro lipid binding assay as previously described [47]. In brief, nitrocellulose membranes pre-spotted with various indicated lipid species (P-6001, P-6002, Echelon, Salt Lake City, UT) were blocked in 3% fat-free BSA for 60 min at room temperature, and then were probed with the purified ApoL6.V6 protein (∼100 ng/ml) in 3% fat-free BSA in TBST for 1 h at room temperature, followed by primary anti-ApoL6 antibody, secondary goat anti-rabbit antibody conjugated with horseradish peroxidase, and ECL detection and imaging.
VIII Statistical Analysis
Unless otherwise stated, data were expressed as mean ± SD. Where applicable, the results were compared by using the unpaired, two-tailed Student t-test, as implemented by Excel 2007 (Microsoft Corp., Redmond, WA). P-value smaller than 0.05 was considered statistically significant.
Results
I Microscopic Analysis Showed that ApoL6 Induces a Mixed Cell Death Phenotype that Includes Both Apoptosis and Necroptosis
DLD1.ApoL6 cells were cultured in three different treatment groups: D.20, D.0.5 and D.0.5 with the addition of 2 anti-apoptotic agents. The drug treatment included Z-VAD (10 µM) and BV-6 (12.5 µM). Z-VAD is a pan-caspase inhibitor, binding to the catalytic site of caspases and thereby inhibiting caspase activities and offering cell protection from apoptosis. BV-6, a SMAC mimetic that antagonizes the cIAP and XIAP, induces necroptosis when the function of caspases, for example, by Z-VAD, is blocked [48]. We observed that Z-VAD+BV-6 indeed offered significant protection and slowed down ApoL6-induced cell death in D.0.5. (Figure 1A) and pushed cell death into a necroptotic phenotype (Figure 1B). Figure 1A showed the average percentage of cell death after 6 hours of treatment. D.20 exhibited a very low (0-5%) cell death as to be expected. In D.0.5 medium alone 50% of cells died. With the addition of Z-VAD and BV-6 cell death was significantly decreased to by half to result in ~25%, indicating that these inhibitors effectively slow the progression of ApoL6 induced cell death.
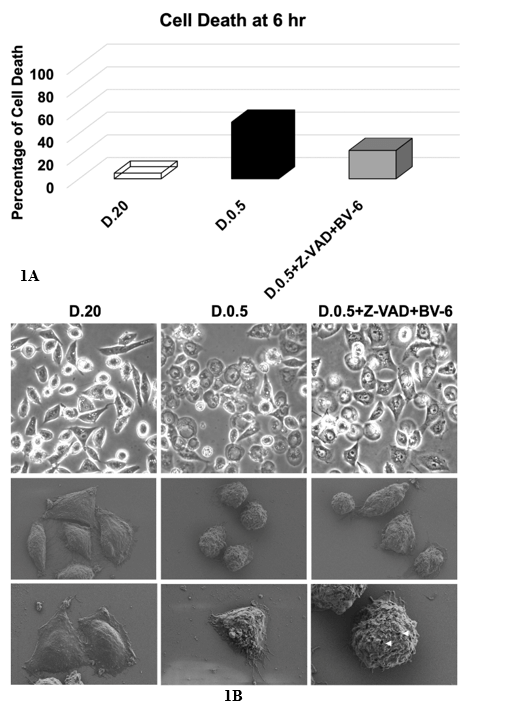
Figure 1: Light Microscopy and Scanning Electron Microscopy of DLD1.ApoL6.
Cells were cultured in three treatment groups: A) D.20 (open column), D.0.5 (black column) and D.0.5 +Z-VAD+BV-6 (grey column) and B) D.20 (left column), D.0.5 (middle column) or D.0.5 +Z-VAD+BV-6 (right column). (A) Cell death was expressed as the percentage of dead cells/total cells. Z-VAD+BV-6 slowed down ApoL6-induced cell death in D.0.5 by an average decrease of 25% over 6 hours of treatment compared to D.0.5 alone. (B) Top- light microscopic images. SEM images were shown in the middle and bottom rows. The middle row offered a perspective of several cells under a lower magnification, while the bottom row was at a higher magnification focused on 1-2 cells. Small membrane holes in necroptotic cells were marked by the white arrows.
We imaged cells using either standard light microscopy (Figure 1B top row) or scanning electron microscopy (SEM) (Figure 1B middle and bottom rows). As previously stated, apoptosis and necroptosis are distinct in their morphological characteristics. Figure 1B displayed the morphology of both pathways. D.20 served as a control of a healthy phenotype, especially in SEM images wherein cells are healthy and large with fully intact membranes. In D.0.5 medium cells proceeded primarily through apoptosis. The representative light microscopy image in the top center panel showed many cells that exhibited cell shrinkage characteristic of apoptosis. This is further visualized in the SEM image (center panel) showing four fixed cells that display apoptotic phenotypes of cell shrinkage and rounding up as well as membrane blebbing [10, 48]. The bottom middle panel showed a more focused and higher magnification of a single cell with clear membrane blebs while still maintaining membrane integrity without rupture.
In drug treated cells (right column) this differed in that representative SEM images cells exhibited the regulated development of small pores associated with plasma membrane rupture in accordance with necroptosis as indicated by the white arrows [48]. SEM of four drug treated cells (right column, middle row) primarily did not have the rounded up and shriveled morphology of apoptotic D.0.5 alone cells but rather showed cellular swelling. It is noteworthy that the left most cell in this image did resemble an apoptotic rounded up phenotype, indicating that both apoptosis and necroptosis are being executed at a 25/75 ratio.
The light microscopy image for drug treated cells also showed a decrease in the number of shriveled cells. The D.0.5 group without drug treatment showed a mixture of both phenotypes, implying that there is a mixture of both apoptosis and necroptosis being executed even in the absence of drug intervention. This feat can be attributed to the release of SMAC upon ApoL6 overexpression as previously established [40]. The release of SMAC promotes necroptosis and in the presence of Z-VAD and BV-6 this push towards necroptosis was enhanced as seen in the right column.
II Western Blotting Analysis Showed Differential Expression of Various Protein Markers
Figure 2 showed the Western blotting images of anti-p-MLKL followed by anti-MLKL and anti-β-actin. β-actin was used as a loading control. It is noteworthy that lane 3 (D.0.5+Z-VAD+BV-6) showed significant decrease in β-actin band intensity. We determined this band to have ~4-fold decrease in intensity relative to D.20. P-MLKL as indicated by the arrow was marked at a molecular weight of approximately 54 kDa. Despite a lower amount of protein loaded into lane 3, there was a prominent signal of p-MLKL, indicating a higher expression of p-MLKL in this treatment. In D.20 the p-MLKL band showed a weak signal compared to that of D.0.5+Z-VAD+BV-6 which is significantly more defined. We determined this to be a roughly 2.5-fold increase in intensity as compared to D.20. Taken together, the 4-fold decrease in protein loaded as indicated by β-actin multiplied by the 2.5x increase in p-MLKL signal displayed a total of ~10-fold increase in p-MLKL expression in D.0.5 +Z-VAD+BV-6. During the execution of necroptosis, MLKL is recruited to the necrosome and is then phosphorylated/activated by RIPK3. The antibody raised against p-MLKL binds to the serine residue at amino acid 345 that becomes phosphorylated in this interaction to activate MLKL. The presence of p-MLKL in all lanes indicates that all samples have the begun the execution of necroptosis to some degree.
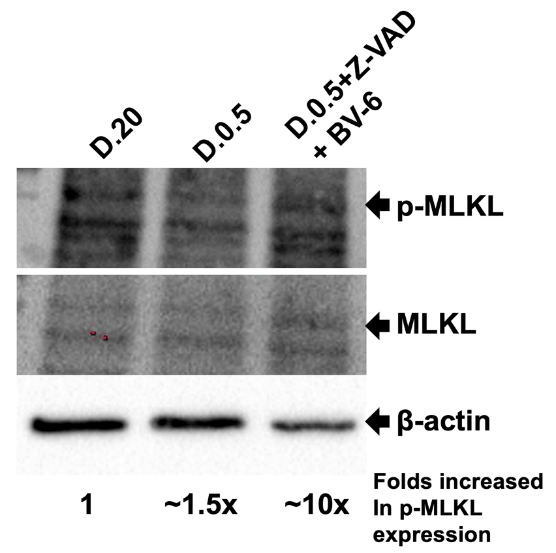
Figure 2: Western Blot Analysis of P-MLKL and MLKL.
Sixty micrograms of intracellular protein extract was added to each lane. Lane 1, Protein sample isolated from cells grown in D.20 (Lane 1), D.0.5 (Lane 2), and D.0.5 +Z-VAD+BV-6 (Lane 3). Membrane was first probed for p-MLKL, then MLKL, and finally β-actin. Respective bands indicated by arrows. The molecular weight of MLKL is 54 kDa and β-actin is 42 kDa. Fold increase in p-MLKL expression was calculated by normalizing band intensity of β-actin and multiplying by the increase in intensity of p-MLKL relative to D.20.
III Immunofluorescence Microscopy Revealed Differential Expression of p-MLKL in Treated Cells
Fluorescence Imaging on an EVOS microscope revealed that in D.0.5 culture conditions (Figure 3A), cells displayed varying levels of p-MLKL expression indicated by the green fluorescence of Alexa 488 secondary antibody. Expression can be divided into three major categories as visible in (c): low p-MLKL shown by relatively low green fluorescence as in box 1, high p-MLKL indicated by bright green fluorescence as in box 2, and no p-MLKL indicated by the absence of any green in box 3. Cell-Mask Deep Red (a) is a plasma membrane specific marker that shows equal expression across all fixed cells.
Cells were also cultured in D.0.5 +Z-VAD+BV-6 followed by fixation, immunofluorescent staining and imaging in the same manner. Figure 3B showed a significant increase in the intensity and frequency of brighter green signal (c), thereby indicating an increase in p-MLKL expression. Consistent with the labelling for (Figure 3A), box 1 represents a lower expression and box 2 represents a strong green signal and p-MLKL expression. It is noteworthy that none of the fixed cells in the drug treated sample expressed absolutely no green, signifying that all these cells have some level of p-MLKL, distinct from D.0.5 alone wherein some cells did not express any detectable levels of p-MLKL.
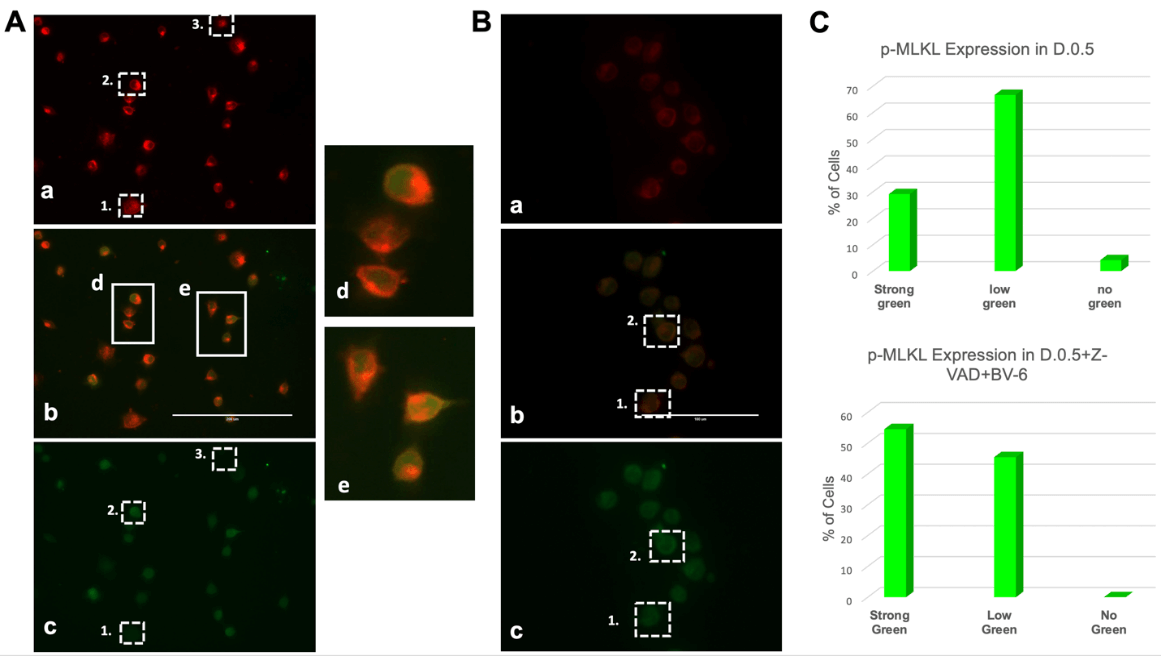
Figure 3: Differential expression of p-MLKL in treated cells.
A) Immunofluorescence Microscopy images generated from DLD1.ApoL6 cultured in D.0.5 media. Plasma membrane marker Cell-Mask Deep Red (a) was expressed equally across all cells, p-MLKL Alexa 488, (c) showed differential expression. Overlap of the two signals (b) represented a distinct yellow ring structure (zoomed image in d/e). Box 1 represented low p-MLKL expression, box 2 represented strong p-MLKL expression and box 3 represented little to no expression. B) displayed the IF images of DLD1.ApoL6 cultured in D.0.5 +Z-VAD+BV-6. Labelling of box 1 and 2 were consistent with that of panel A. Scale was 200 μm for panel A and 100 μm for panel B. C) Quantification of cells in each phenotype was expressed by the bar graphs for each treatment group. Total cells were counted and number in each phenotypic group presented as a percentage as indicated on the y-axis.
Furthermore, the overlap of Cell-Mask Deep Red and Alexa 488 in (b) for both panel A and B showed a colocalization of p-MLKL to the plasma membrane. The overlap of green and red caused a distinct yellow/orange ring-like structure. This ring-like structure was most clearly distinguished in (Figure 3A) boxes d and e wherein the overlap of p-MLKL and Cell-Mask Deep Red showed a significant amount of p-MLKL co-localized to the plasma membrane.
Quantification of the number of cells in each phenotype for the two different treatment groups was expressed as a percentage in the bar graphs of (Figure 3C). In D.0.5, 67% of fixed cells had a low level of p-MLKL, 29% showed strong green signal and expression and 4% had absolutely no expression of p-MLKL. In D.0.5 +Z-VAD+BV-6, 45% of cells showed a low level of expression and 55% displayed high levels of expression. The presence of multiple phenotypes displayed a mixture of cell death pathways, which directly correlates to our initial hypothesis that ApoL6 is also capable of inducing necroptosis, even without drug intervention. Again, this can be linked to the release of SMAC upon ApoL6 overexpression as previously established. The increase in frequency of high p-MLKL expression for the drug treated group indicated an enhanced pushed towards necroptosis, in agreement with the results of (Figure 2).
IV Overexpression of ApoL6 Promotes Extracellular Release of IL-1β
Via the use of an ELISA kit as described, we measured the absorbance at 450 nm and converted each measurement into [IL-1β] released. Seven standard concentrations ranging from 0 pg/mL to 250 pg/mL were also included in the data set (data not shown) to generate a standard curve and establish blank absorbance at 0 pg/mL. D.20 served as a baseline for comparison. The increase in concentration relative to D.20 for D.0.5 alone and D.0.5 +Z-VAD+BV-6 was indicated as percent increase in (Figure 4).
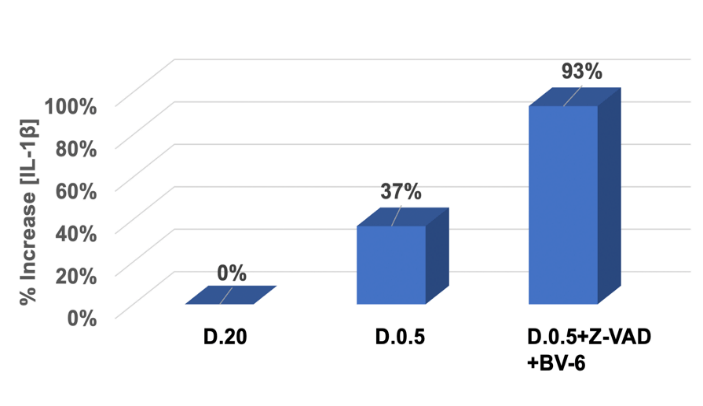
Figure 4: ApoL6 induces IL-1β release during cell death.
ELISA assay was used to determine the percent change in IL-1β released to the extracellular space in indicated treatments. The y-axis represented percent change in concentration relative to D.20 control. The x- axis indicated various treatment conditions. D.0.5 showed a 37% increase relative to D.20 while D.0.5 in the presence of Z-VAD and BV-6 showed a significant increase of 93 %.Data analysis included the generation of a standard curve (data not shown) and the subtraction of a blank absorbance at 0 pg/mL from all samples.
Percent increase relative to D.20 was determined by the following equation: (A450 of D.0.5 with or without drugs - A450 of D.20) / A450 of D.20. In D.0.5 there was a 37% increase in [IL-1β] released into the medium relative to D.20, corresponding to an increase in the execution of the necroptotic pathway. As expected, in the presence of apoptotic inhibitors Z-VAD and BV-6, there was a substantial increase of IL-1β release. This sample group showed a 93% increase of IL-1β release relative to D.20 control, displaying a prominent induction of necroptotic cell death. When compared to the D.0.5 samples in the absence of drug treatment, there was still a 41% increase when Z-VAD and BV-6 were added, corresponding to the enhanced push towards necroptosis. This data further displays an ability to manipulate the pathway of cell death and is an indication that ApoL6 is capable of inducing necroptosis in agreement with the results of (Figures 1-3).
V ApoL6 Binds Unique PIPs
By a protein-lipid overlay binding assay, (Figure 5) we showed that ApoL6 is a phospholipid binding protein that interacts effectively and specifically displays strong binding to the following phospholipids: PI4P, PI5P, PI(4, 5)P2, PI(3, 4, 5)P3 , PI3P, and PI(3, 5)P2. Importantly, PI(4, 5)P2, PI4P and PI5P that showed strong interactions with ApoL6 have been previously shown to be present on the plasma membrane and preferentially bound by MLKL to promote membrane permeabilization [31, 32].

Figure 5: ApoL6 binds strongly with PIPs.
A protein-lipid overlay assay showed ApoL6 bound with a variety of PIPs and other phospholipid species. ApoL6 appeared to overall interact effectively and specifically displays strong binding to the following PIPs: PI4P, PI(4,5)P2, PI5P, PI(3,4,5)P3 , PI3P, and PI(3,5) P2.
Lipid abbreviations are as follows: TG (triglyceride), PI (phosphatidylinositol), DAG (diacylglycerol), PI4P (PtdIns(4)P), PA (phosphatidic acid), PI4,5P2 (PtdIns(4,5)P2), PS (phosphatidylserine), PI3,4,5P3 (PtsIns(3,4,5)P3), PE (phosphatidylethanolamine), CHO (cholesterol), PC (phosphatidylcholine), SM (sphingomyelin), PG (phosphatidylglycerol), 3-SGCer (3-sulfoglactoslyceramide (Sulfatide)), CL (cardiolipin), S1P (shingosine-1-phosphate), LPA (lysophosphatidic acid), LPC (Lysophosphatidylcholine), PI3,4P2 (PtdIns(3,4)P2), PI3,5P2 (PtdIns(3,5)P2), PI3P (PtdIns(3)P), PI5P (PtdIns(5)P), and Solvent blank (Blank).
Discussion
In this study, we showed that under various controlled conditions, ApoL6 can induce predominate apoptosis or necroptosis phenotypes. Without apoptosis inhibitors, a mixed population of dead cells includes majority apoptotic and smaller levels of necroptotic phenotypes. When drug treatment of Z-VAD and BV-6 were added, ApoL6-induced cell death was switched from primarily apoptosis to necroptosis. Figures 1-4 showed that in the absence of apoptotic inhibitors, DLD1.ApoL6 can still induce a mixed phenotype. This is in agreement with our previous work establishing that upon ApoL6 overexpression SMAC is released from the mitochondria [40]. With the addition of Z-VAD to block caspases and BV-6 SMAC mimetic, the push towards necroptosis is enhanced and cells primarily proceed via this path. The previous results further validate what we observed here in different cellular systems including adenovirus ApoL6 overexpression. Thus, in this study we expanded on the role of ApoL6 in regulated cell death to include both apoptosis and necroptosis. Although we mainly used DLD1.ApoL6 cell as the model to dissect the role of ApoL6 in RCD, as in our previously published work, we have used other cell models where we could ectopically express ApoL6, for example, DU-145 (prostate), HEK 293 (human embryonic kidney), and LDCs (human atherosclerotic lesion derived cells) [40, 41].
Light and SEM images (Figure 1B) showed a mixture of the morphological characteristics of apoptosis and necroptosis. Apoptosis was primarily identified by cell shriveling, rounding up and membrane blebbing. Necroptosis was marked by the selective formation of small holes in the plasma membrane and cellular swelling. Membrane rupture was not the primary defining feature of necroptotic death in our setting due to SEM imaging occurring only on cells that were still attached to the coverslip and then fixed. Cells that had proceeded all the way through the necroptotic path that would exhibit significant membrane rupture would likely be in the medium rather than remaining attached. Some of the imaged cells that remained attached were the ones still undergoing the process of necroptosis as indicated by the regulated production of small membrane pores.
Figures 2 & 3 examined levels of the intracellular necroptotic marker p-MLKL. We found that upon overexpression of ApoL6 in DLD1, there was an increase in p-MLKL levels. In the presence of Z-VAD and BV-6, levels of p-MLKL were further enhanced and co-localized to the plasma membrane.
We showed that under different treatment conditions, varying levels of IL-1β are released by DLD1.ApoL6 (Figure 4). Under D.20 or normal media conditions, minimal amounts of IL-1β were released but under induction condition with or without apoptosis inhibitors there was a substantial increase of 93% and 37% respectively. The dramatic difference in IL-1β levels is a strong indicator that the pathway of cell death can be manipulated upon ApoL6 overexpression. This data further supports our initial hypothesis that ApoL6 can induce a mixed cell death phenotype and that ApoL6-initiated necroptotic cells release substantially more IL-1β. Our light microscopy, Western Blotting and immunofluorescence microscopy pertained to the examination of necroptosis being executed from an intracellular perspective. The release of IL-1β provides validation that necroptosis had been induced from a downstream and extracellular perspective.
Using the protein-lipid overlay assay, we demonstrated that ApoL6, like its closely related protein ApoL1, is a phospholipid binding protein, with strong interactions with PIPs [47]. PIPs play critical roles in regulating cellular signaling pathways. The homeostatic equilibrium of PIPs determines cellular proliferation, differentiation, trafficking, and RCD [49]. Previously we have shown that ApoL1, an inducer of autophagy-associated cell death, displays strong binding to a different set of lipids including phosphatidic acid (PA) and cardiolipin (CL), some unique phospholipid species to autophagic regulation [47]. ApoL6 and ApoL1 both possess the ability to bind different and unique lipids indicating that ApoL6 appears overall to bind more to PIPs that are present on the plasma membrane.
Intriguingly, PI(4, 5)P2, PI4P and PI5P that show strong binding to ApoL6 in our assay are the unique PIPs which have been previously shown to be present on the plasma membrane and preferentially bound by MLKL to promote membrane permeabilization [31, 32]. For example, it has been demonstrated that the p-MLKL forms an oligomer that binds to PIPs and cardiolipin. This property allows p-MLKL to move from the cytosol to the plasma and intracellular membranes, where it directly disrupts membrane integrity, resulting in necroptosis [32]. Wang et al. also demonstrated preferential binding to membrane PIPs such as PI(4, 5)P2 and PI4P as determined by the lipid overlay assay similar to the one described in this study [32]. In addition, Dondelinger et al. showed that the N-terminal four-helical bundle domain (4HBD) of MLKL is required and sufficient to induce MLKL oligomerization [31]. They also found that a patch of amino acids that harbor positive charge on the surface of the 4HBD binds to PIPs and allows recruitment of MLKL to the plasma membrane. Oligomerized p-MLKL induces necroptosis by directly permeabilizing the plasma membrane. Importantly, inhibiting the formation of PI(5)P and PI(4,5)P2 specifically blocks tumor necrosis factor (TNF)-mediated necroptosis but not apoptosis [31].
Furthermore, Quarato et al. recently showed that 4HNB preferentially binds to phosphorylated inositol polar head groups of PI(4,5)P2, and at the membrane, the 4HNB undergoes a "rolling over" mechanism to expose additional higher-affinity PIP-binding sites responsible for robust association to the membrane, leading to the membrane disruption [50]. However, while functions of PIPs have been extensively studied, the mechanisms by which PIPs are delivered to the plasma membrane are poorly understood. It is reasonable to speculate that ApoL6 plays a role in the binding and trafficking intracellular PIPs to the plasma membrane where PIPs subsequently bind with oligomerized p-MLKL. In particular, ApoL6 possesses higher affinity to PI(4, 5)P2, PI4P, and PI5P, the MLKL preferred binding partners as determined by Wang et al. and Dondlinger et al. [31, 32]. Our results suggest that overexpression of ApoL6 under a specific cellular context, for example, caspase inactivation, can lead to the induction of necroptosis, in part, by its intracellular binding of PIPs. This would be a critical and newly identified function in necroptosis because PIPs must be present on the membrane in order for p-MLKL to form pores and permeabilize the membrane to proceed with downstream events as associated with necroptosis.
It is also logical to speculate that the level of ApoL6 expression can dictate which pathway by which a cell proceeds to die. In a non-synchronized cell culture condition, cells with the higher expression of ApoL6, the more likely that necroptosis would be the preferred pathway and lower expression correlating to an enhanced push towards apoptosis. There is the possibility of a molecular switch that could be playing a role in this decision between apoptosis and necroptosis related to ApoL6 expression. With the new information presented in this study that ApoL6 induces necroptosis, further investigation can be done to examine the role of ApoL6 in necroptosis including PIP trafficking and the mechanism by which a cell makes the decision of which cell death pathway to execute.
Acknowledgements
The study is, in part, supported by 2 pilot projects of UNMHSC CTSA center grant (1UL1RR031977-01). We thank Ms. Tamara Howard for her excellent SEM service at the EM Core Facility, University of New Mexico School of Medicine.
Abbreviations
ApoL6: Apolipoprotein L6
MLKL: Mixed Lineage Kinase Domain-Like Protein
IL-1β: Interleukin-1Beta
Article Info
Article Type
Research ArticlePublication history
Received: Fri 12, Jun 2020Accepted: Sat 11, Jul 2020
Published: Tue 21, Jul 2020
Copyright
© 2023 Chien-An Andy Hu. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited. Hosting by Science Repository.DOI: 10.31487/j.COR.2020.07.12
Figures & Tables
Cells were cultured in three treatment groups: A) D.20 (open column), D.0.5 (black column) and D.0.5 +Z-VAD+BV-6 (grey column) and B) D.20 (left column), D.0.5 (middle column) or D.0.5 +Z-VAD+BV-6 (right column). (A) Cell death was expressed as the percentage of dead cells/total cells. Z-VAD+BV-6 slowed down ApoL6-induced cell death in D.0.5 by an average decrease of 25% over 6 hours of treatment compared to D.0.5 alone. (B) Top- light microscopic images. SEM images were shown in the middle and bottom rows. The middle row offered a perspective of several cells under a lower magnification, while the bottom row was at a higher magnification focused on 1-2 cells. Small membrane holes in necroptotic cells were marked by the white arrows.
Sixty micrograms of intracellular protein extract was added to each lane. Lane 1, Protein sample isolated from cells grown in D.20 (Lane 1), D.0.5 (Lane 2), and D.0.5 +Z-VAD+BV-6 (Lane 3). Membrane was first probed for p-MLKL, then MLKL, and finally β-actin. Respective bands indicated by arrows. The molecular weight of MLKL is 54 kDa and β-actin is 42 kDa. Fold increase in p-MLKL expression was calculated by normalizing band intensity of β-actin and multiplying by the increase in intensity of p-MLKL relative to D.20.
A) Immunofluorescence Microscopy images generated from DLD1.ApoL6 cultured in D.0.5 media. Plasma membrane marker Cell-Mask Deep Red (a) was expressed equally across all cells, p-MLKL Alexa 488, (c) showed differential expression. Overlap of the two signals (b) represented a distinct yellow ring structure (zoomed image in d/e). Box 1 represented low p-MLKL expression, box 2 represented strong p-MLKL expression and box 3 represented little to no expression. B) displayed the IF images of DLD1.ApoL6 cultured in D.0.5 +Z-VAD+BV-6. Labelling of box 1 and 2 were consistent with that of panel A. Scale was 200 μm for panel A and 100 μm for panel B. C) Quantification of cells in each phenotype was expressed by the bar graphs for each treatment group. Total cells were counted and number in each phenotypic group presented as a percentage as indicated on the y-axis.
ELISA assay was used to determine the percent change in IL-1β released to the extracellular space in indicated treatments. The y-axis represented percent change in concentration relative to D.20 control. The x- axis indicated various treatment conditions. D.0.5 showed a 37% increase relative to D.20 while D.0.5 in the presence of Z-VAD and BV-6 showed a significant increase of 93 %.Data analysis included the generation of a standard curve (data not shown) and the subtraction of a blank absorbance at 0 pg/mL from all samples.
A protein-lipid overlay assay showed ApoL6 bound with a variety of PIPs and other phospholipid species. ApoL6 appeared to overall interact effectively and specifically displays strong binding to the following PIPs: PI4P, PI(4,5)P2, PI5P, PI(3,4,5)P3 , PI3P, and PI(3,5) P2.
Lipid abbreviations are as follows: TG (triglyceride), PI (phosphatidylinositol), DAG (diacylglycerol), PI4P (PtdIns(4)P), PA (phosphatidic acid), PI4,5P2 (PtdIns(4,5)P2), PS (phosphatidylserine), PI3,4,5P3 (PtsIns(3,4,5)P3), PE (phosphatidylethanolamine), CHO (cholesterol), PC (phosphatidylcholine), SM (sphingomyelin), PG (phosphatidylglycerol), 3-SGCer (3-sulfoglactoslyceramide (Sulfatide)), CL (cardiolipin), S1P (shingosine-1-phosphate), LPA (lysophosphatidic acid), LPC (Lysophosphatidylcholine), PI3,4P2 (PtdIns(3,4)P2), PI3,5P2 (PtdIns(3,5)P2), PI3P (PtdIns(3)P), PI5P (PtdIns(5)P), and Solvent blank (Blank).
References
- M O Hengartner (2000) The biochemistry of apoptosis. Nature 407: 770-776. [Crossref]
- B Vogelstein, D Lane, A J Levine (2000) Surfing the p53 network. Nature 408: 307-310. [Crossref]
- Richard A Lockshin, Zahra Zakeri (2007) Cell death in health and disease. J Cell Mol Med 11: 1214-1224. [Crossref]
- Yaron Fuchs, Hermann Steller (2015) Live to die another way: modes of programmed cell death and the signals emanating from dying cells. Nat Rev Mol Cell Biol 16: 329-344. [Crossref]
- Swapna A Gudipaty, Christopher M Conner, Jody Rosenblatt, Denise J Montell (2018) Unconventional Ways to Live and Die: Cell Death and Survival in Development, Homeostasis, and Disease. Annu Rev Cell Dev Biol 34: 311-332. [Crossref]
- Ricardo Weinlich, Andrew Oberst, Christopher P Dillon, Laura J Janke, Sandra Milasta et al. (2013) Protective roles for caspase-8 and cFLIP in adult homeostasis. Cell Rep 5: 340-348. [Crossref]
- Alessandro Annibaldi, Pascal Meier (2018) Checkpoints in TNF-Induced Cell Death: Implications in Inflammation and Cancer. Trends Mol Med 24: 49-65. [Crossref]
- J F Kerr, A H Wyllie, A R Currie (1972) Apoptosis: a basic biological phenomenon with wide-ranging implications in tissue kinetics. Br J Cancer 26: 239-257. [Crossref]
- Lorenzo Galluzzi, Alejandro López Soto, Sharad Kumar, Guido Kroemer (2016) Caspases Connect Cell-Death Signaling to Organismal Homeostasis. Immunity 44: 221-231. [Crossref]
- Susan Elmore (2007) Apoptosis: a review of programmed cell death. Toxicol Pathol 35: 495-516. [Crossref]
- C DE DUVE (1963) The lysosome. Sci Am 208: 64-72. [Crossref]
- Shigeomi Shimizu, Shinya Honda, Satoko Arakawa, Hirofumi Yamaguchi (2014) Alternative macroautophagy and mitophagy. Int J Biochem Cell Biol 50: 64-66. [Crossref]
- Beth Levine, Guido Kroemer (2019) Biological Functions of Autophagy Genes: A Disease Perspective. Cell 176: 11-42. [Crossref]
- B T Cookson, M A Brennan (2001) Pro-inflammatory programmed cell death. Trends Microbiol 9: 113-114. [Crossref]
- Susan L Fink, Brad T Cookson (2007) Pyroptosis and host cell death responses during Salmonella infection. Cell Microbiol 9: 2562-2570. [Crossref]
- Alexei Degterev, Zhihong Huang, Michael Boyce, Yaqiao Li, Prakash Jagtap et al. (2005) Chemical inhibitor of nonapoptotic cell death with therapeutic potential for ischemic brain injury. Nat Chem Biol 1: 112-119. [Crossref]
- Ricardo Weinlich, Andrew Oberst, Helen M Beere, Douglas R Green (2017) Necroptosis in development, inflammation and disease. Nat Rev Mol Cell Biol 18: 127-136. [Crossref]
- Lorenzo Galluzzi, Oliver Kepp, Francis Ka Ming Chan, Guido Kroemer (2017) Necroptosis: Mechanisms and Relevance to Disease. Annu Rev Pathol 12: 103-130. [Crossref]
- Scott J Dixon, Kathryn M Lemberg, Michael R Lamprecht, Rachid Skouta, Eleina M Zaitsev et al. (2012) Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell 149: 1060-1072. [Crossref]
- Amy Tarangelo, Leslie Magtanong, Kathryn T Bieging Rolett, Yang Li, Jiangbin Ye et al. (2018) p53 Suppresses Metabolic Stress-Induced Ferroptosis in Cancer Cells. Cell Rep 22: 569-575. [Crossref]
- Leslie Magtanong, Scott J Dixon (2018) Ferroptosis and Brain Injury. Dev Neurosci 40: 382-395. [Crossref]
- N Holler, R Zaru, O Micheau, M Thome, A Attinger et al. (2000) Fas triggers an alternative, caspase-8-independent cell death pathway using the kinase RIP as effector molecule. Nat Immunol 1: 489-495. [Crossref]
- Peter Vandenabeele, Lorenzo Galluzzi, Tom Vanden Berghe, Guido Kroemer (2010) Molecular mechanisms of necroptosis: an ordered cellular explosion. Nat Rev Mol Cell Biol 11: 700-714. [Crossref]
- Alexei Degterev, Junichi Hitomi, Megan Germscheid, Irene L Ch'en, Olga Korkina et al. (2008) Identification of RIP1 kinase as a specific cellular target of necrostatins. Nat Chem Biol 4: 313-321. [Crossref]
- Young Sik Cho, Sreerupa Challa, David Moquin, Ryan Genga, Tathagat Dutta Ray et al. (2009) Phosphorylation-driven assembly of the RIP1-RIP3 complex regulates programmed necrosis and virus-induced inflammation. Cell 137: 1112-1123. [Crossref]
- Sudan He, Lai Wang, Lin Miao, Tao Wang, Fenghe Du et al. (2009) Receptor interacting protein kinase-3 determines cellular necrotic response to TNF-alpha. Cell 137: 1100-1111. [Crossref]
- Duan Wu Zhang, Jing Shao, Juan Lin, Na Zhang, Bao Ju Lu et al. (2009) RIP3, an energy metabolism regulator that switches TNF-induced cell death from apoptosis to necrosis. Science 325: 332-336. [Crossref]
- Liming Sun, Huayi Wang, Zhigao Wang, Sudan He, She Chen et al. (2012) Mixed lineage kinase domain-like protein mediates necrosis signaling downstream of RIP3 kinase. Cell 148: 213-227. [Crossref]
- Jie Zhao, Siriporn Jitkaew, Zhenyu Cai, Swati Choksi, Qiuning Li et al. (2012) Mixed lineage kinase domain-like is a key receptor interacting protein 3 downstream component of TNF-induced necrosis. Proc Natl Acad Sci U S A 109: 5322-5327. [Crossref]
- Jixi Li, Thomas McQuade, Ansgar B Siemer, Johanna Napetschnig, Kenta Moriwaki et al. (2012) The RIP1/RIP3 necrosome forms a functional amyloid signaling complex required for programmed necrosis. Cell 150: 339-350. [Crossref]
- Yves Dondelinger, Wim Declercq, Sylvie Montessuit, Ria Roelandt, Amanda Goncalves et al. (2014) MLKL compromises plasma membrane integrity by binding to phosphatidylinositol phosphates. Cell Rep 7: 971-981. [Crossref]
- Huayi Wang, Liming Sun, Lijing Su, Josep Rizo, Lei Liu et al. (2014) Mixed lineage kinase domain-like protein MLKL causes necrotic membrane disruption upon phosphorylation by RIP3. Mol Cell 54: 133-146. [Crossref]
- Agnieszka Kaczmarek, Peter Vandenabeele, Dmitri V Krysko (2013) Necroptosis: the release of damage-associated molecular patterns and its physiological relevance. Immunity 38: 209-223. [Crossref]
- Manolis Pasparakis, Peter Vandenabeele (2015) Necroptosis and its role in inflammation. Nature 517: 311-320. [Crossref]
- Seamus J Martin (2016) Cell death and inflammation: the case for IL-1 family cytokines as the canonical DAMPs of the immune system. FEBS J 283: 2599-2615. [Crossref]
- Gloria Lopez Castejon, David Brough (2011) Understanding the mechanism of IL-1β secretion. Cytokine Growth Factor Rev 22: 189-195. [Crossref]
- James E Vince, W Wei Lynn Wong, Ian Gentle, Kate E Lawlor, Ramanjaneyulu Allam et al. (2012) Inhibitor of apoptosis proteins limit RIP3 kinase-dependent interleukin-1 activation. Immunity 36: 215-227. [Crossref]
- Monica Yabal, Philipp J Jost (2014) XIAP as a regulator of inflammatory cell death: the TNF and RIP3 angle. Mol Cell Oncol 2: e964622. [Crossref]
- K Moriwaki, F K M Chan (2017) The Inflammatory Signal Adaptor RIPK3: Functions Beyond Necroptosis. Int Rev Cell Mol Biol 328: 253-275. [Crossref]
- Zhihe Liu, Huimei Lu, Zeyu Jiang, Andrzej Pastuszyn, Chien an A Hu (2005) Apolipoprotein L6, a novel proapoptotic Bcl-2 homology 3-only protein, induces mitochondria-mediated apoptosis in cancer cells. Mol Cancer Res 3: 21-31. [Crossref]
- Zhaorigetu S, Yang Z, Toma I, McCaffrey TA, Hu CA (2011) Apolipoprotein L6, induced in atherosclerotic lesions, promotes apoptosis and blocks Beclin 1-dependent autophagy in atherosclerotic cells. J Biol Chem 286: 27389-27398. [Crossref]
- D C Huang, A Strasser (2000) BH3-Only proteins-essential initiators of apoptotic cell death. Cell 103: 839-842. [Crossref]
- Suzanne Cory, Jerry M Adams (2002) The Bcl2 family: regulators of the cellular life-or-death switch. Nat Rev Cancer 2: 647-656. [Crossref]
- Ya Tan, Mailin Gan, Yuan Fan, Liang Li, Zhijun Zhong et al. (2019) miR-10b-5p regulates 3T3-L1 cells differentiation by targeting Apol6. Gene 687: 39-46. [Crossref]
- J Yu, L Zhang, P M Hwang, C Rago, K W Kinzler et al. (1999) Identification and classification of p53-regulated genes. Proc Natl Acad Sci U S A 96: 14517-14522. [Crossref]
- T Murakami, Z L Song, H Hinenoya, A Ohtsuka, T Taguchi et al. (1987) Lysine-mediated tissue osmication in combination with a tannin-osmium conductive staining method for non-coated scanning electron microscopy of biological specimens. Arch Histol Jpn 50: 485-493. [Crossref]
- Guanghua Wan, Siqin Zhaorigetu, Zhihe Liu, Ramesh Kaini, Zeyu Jiang et al. (2008) Apolipoprotein L1, a novel Bcl-2 homology domain 3-only lipid-binding protein, induces autophagic cell death. J Biol Chem 283: 21540-21549. [Crossref]
- Yingying Zhang, Xin Chen, Cyril Gueydan, Jiahuai Han (2018) Plasma membrane changes during programmed cell deaths. Cell Res 28: 9-21. [Crossref]
- Matthias P Wymann, Roger Schneiter (2008) Lipid signalling in disease. Nat Rev Mol Cell Biol 9: 162-176. [Crossref]
- Giovanni Quarato, Cliff S Guy, Christy R Grace, Fabien Llambi, Amanda Nourse et al. (2016) Sequential Engagement of Distinct MLKL Phosphatidylinositol-Binding Sites Executes Necroptosis. Mol Cell 61: 589-601. [Crossref]