An Almost Fatal Case of the Hypereosinophilic Syndrome: A Dilemma Between Vasculitis or Clonal Disorder
A B S T R A C T
Hypereosinophilic syndrome (HES) is a rare and potentially life-threatening disorder. While many conditions can cause eosinophilia, it can also be provoked by rarer diseases such as vasculitis or myeloproliferative disorders. Here we present an almost fatal presentation of HES characterized with both vasculitis and clonal features. Treatment with combined chemo-immunotherapy allowed reversal of most of the complications. There is still much uncertainty in the diagnosis and management of HES, and research should be done to improve knowledge on pathophysiology and cytogenetics of this disease in order to improve treatment and prevent life-threatening complications.
Introduction
Hypereosinophilic syndrome (HES) is a rare and underdiagnosed entity that has been redefined in the last few years.1,2 However, it still contains a large area of uncertainty in defining both exact diagnosis and management.2 Here we present a case of steroid-unresponsive HES with some components of vasculitis, that eventually partially reversed under combined chemo-immunotherapy.
Case Report
A 41-year-old woman presented to the emergency department with constitutional symptoms and diffuse arthralgias waxing and waning for a few weeks. On physical examination, she had no fever, vital signs were normal, with no signs of arthritis and no rash. Cardiac examination revealed a holosystolic murmur, and nail examination revealed splinter hemorrhages (Figure 1A). Electrocardiogram (ECG) and chest x-ray (CXR) were normal. Laboratory results revealed a leukocyte count of 10.2x109/L with 28% eosinophils, normal hemoglobin and platelet count, elevated C-reactive protein (50 mg/L), creatinine phosphokinase (CPK) (333 U/L) and lactate dehydrogenase (LDH) (733 U/L). Liver function tests, erythrocyte sedimentation rate, renal function tests and electrolytes were normal.
Previous eosinophil counts 6 months pre-hospitalization were within normal range. She did not start using new drugs or chemicals in the last period, she had no respiratory/sinuses or gastrointestinal clinical findings. Infectious workup including serologies for HIV, Toxocara, Trichinella and Strongyloides, blood and urinary cultures and fecal examination for parasites was negative, and she did not improve under a one-week course of doxycycline. Total body CT (including CT angiography) revealed no sign of malignancy, vasculitis, sinusitis or pulmonary involvement. Immunologic workup revealed positive antinuclear antibody (speckled ++) with negative anti-dsDNA, extractable nuclear antigen screen including detailing, antineutrophilic cytoplasmic antibody (ANCA) including myeloperoxidase and proteinase 3, direct coombs, complement, anticardiolipin and anti-beta-2-glycopreotein-I antibodies but with positive lupus anticoagulant. Nail capillaroscopy revealed signs consistent with vasculitis: bleeding, slow capillary flow, branched and thrombotic capillaries and neo-angiogenesis. Ophthalmologic examination did not demonstrate any sign of vasculitis but showed pathological Shirmer test (without clinically dry eyes). Bone scintigraphy was normal. Transthoracic echocardiography showed moderate-severe pulmonary hypertension, moderate mitral regurgitation (MR) and suspected vegetation.
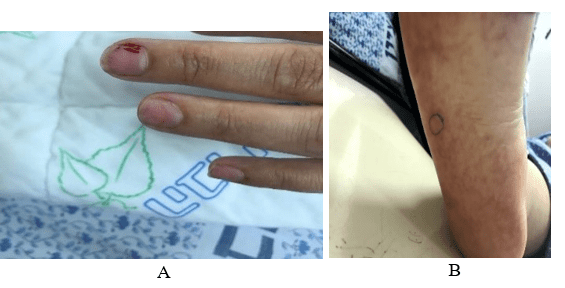
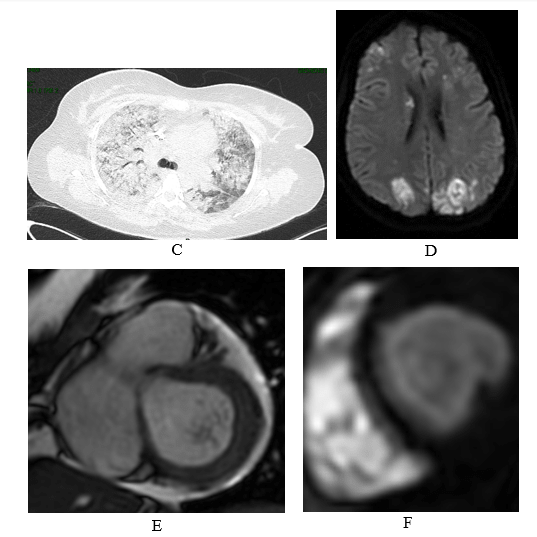
Figure 1: Patient’s clinical and radiologic findings.
A) Splinter hemorrhages.
B) Unspecified rash.
C) Chest CT showing bilateral ground glass opacities suspicious for alveolar hemorrhage.
D) Brain MRI showing occipital infarcts with microhemorrhages.
E) Cardiac MRI showing multiple left ventricular thrombi.
F) Cardiac MRI showing diffuse subendocardial delayed enhancement in a distribution not fitting to a coronary territory.
The negative blood cultures and partially positive immunologic workup led to suspicion of non-bacterial thrombotic endocarditis, but transesophageal echocardiography ruled out this possibility, although it demonstrated marked thickening of apico-posterior lateral walls. Workup for clonal eosinophilia including research for PDGFR-α, BCR-ABL, c-kit and JAK-2 rearrangements was negative. Bone marrow aspiration revealed normal cellularity with no excess of blasts, increased eosinophil counts (22%) with normal morphology and no mast cells. Flow cytometry revealed no eosinophil or lymphocytic clonality, T cell rearrangement did not demonstrate clonality and cytogenetic examination was normal. Bone marrow biopsy showed infiltration with normal eosinophils (22%) with no mast cells.
By this stage, we concluded as hypereosinophilia of unknown origin with some findings of vasculitis, and high-dose glucocorticoid (methylprednisolone 1 g/day) pulse was started. After 5 days of treatment, no improvement in the eosinophil counts was observed and signs of end-organ damages started to appear (Figure 2):
• Dermatologic involvement: a non-specific rash appeared on lower extremities and skin biopsy revealed non-specific findings without vasculitis or eosinophilic infiltration (Figure 1B).
• Cardiac involvement: The patient reported chest pain and dyspnea. Blood analyses demonstrated elevation in high-sensitive troponin (0.5 ng/mL), as well as NT-pro-BNP (10.000 pg/mL). ECG showed sinus tachycardia with ST/T changes in the anterolateral walls. Cardiac MRI showed signs of stage 2 eosinophilic endo-myocarditis: diffuse subendocardial infiltration including in trabecula, with small thrombi and moderate-severe MR (Figure 1E-1F).
• Neurologic involvement: Hand paresis appeared during the disease and progressed to complete left hemiparesis accompanied by cortical blindness. Brain CT and MRI revealed brain infarcts in parietal and occipital territories, with secondary bleeding (Figure 1D).
• Lung involvement: The patient then developed hemoptysis and dyspnea. Chest CT revealed diffuse ground-glass opacities suspected for alveolar hemorrhage (Figure 1C) and blood tests showed decrease in hemoglobin. Due to respiratory distress she was transferred to the intensive care unit where she was intubated and mechanically ventilated.
By this time, the case was concluded as steroid-resistant HES with vasculitis components and life-threatening organ involvement. Anticoagulant therapy was added due to hypercoagulable state, and wide range anti-eosinophilic therapy, targeting possible etiologies, was intensified by adding high-dose hydroxyurea (4.5 g/day), imatinib (400 mg/day), intravenous cyclophosphamide (500 mg) and subcutaneous mepolizumab (300 mg), while continuing high-dose glucocorticoid therapy. Under this treatment, eosinophil counts decreased significantly (30 to 0.49 x 109/L) while severe pancytopenia developed. Imatinib, hydroxyurea, cyclophosphamide and anticoagulation were therefore discontinued, and plasma exchange therapy was started, while maintaining both glucocorticoids and mepolizumab. After a total of 11 plasma exchange therapies, a slow clinical improvement was observed, while eosinophil count did not rise. Anticoagulation, low-dose hydroxyurea (500 mg/day) and one in two weeks cyclophosphamide (500 mg) were re-introduced after resolving of pancytopenia, glucocorticoids were slowly tapered down, mepolizumab was continued once in three weeks, and plasma exchange was replaced by monthly intravenous immunoglobulins.
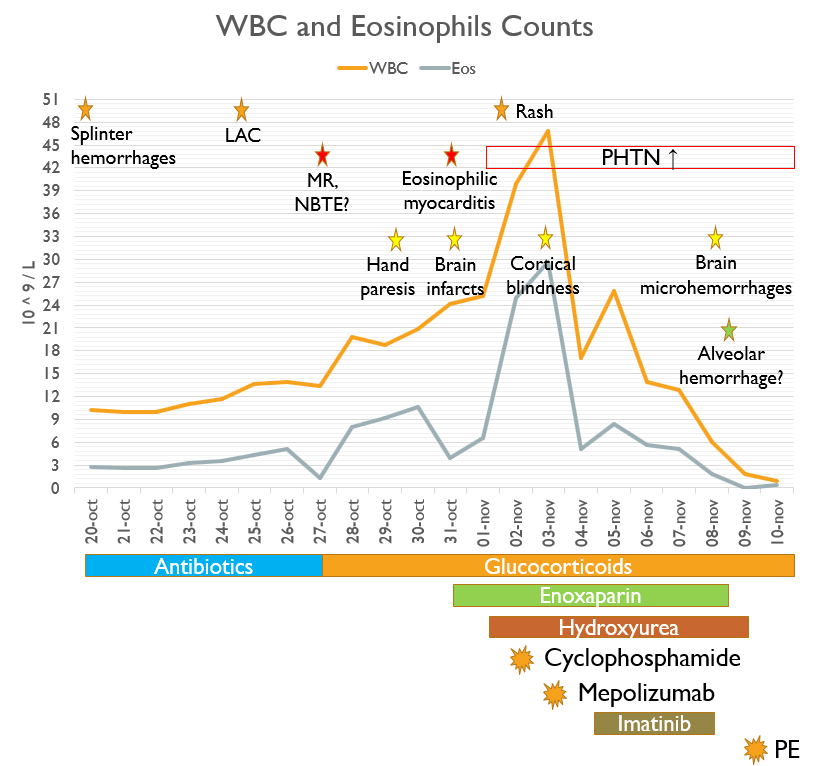
Figure 2: Clinical, laboratory and imaging course and different treatments used.
Eos: Eosinophils; LAC: Lupus anticoagulant; MR: Mitral regurgitation; NBTE: Non-bacterial thrombotic endocarditis; PE: Plasma exchange; PHTN: Pulmonary hypertension; WBC: White blood cells.
Clinically, patient's vision, complete respiratory function and partial neurological motor function recovered. Inflammatory markers, LDH and CPK normalized. High-sensitive troponin and NT-pro-BNP normalized as well, but cardiac function was still impaired: follow-up echocardiography showed restrictive cardiomyopathy with moderate MR and improved pulmonary hypertension. Clinically, she still presented with high blood pressure and recurrent events of pulmonary congestion that were controlled by medications without need for mitral valve replacement. Both non-specific rash and splinter hemorrhages disappeared, and repeated lupus anticoagulant (3 months after the first test) was negative. On follow-up examination 6 months later, the patient was still in clinical and laboratory remission under maintenance therapy as described earlier.
Discussion
HES is a complex syndrome that is divided between clonal, reactive and idiopathic etiologies [1, 2]. Clonal eosinophilia includes all myeloproliferative disorders that may present with eosinophilia (myeloid variant HES or M-HES) and are defined either as cytogenetic changes or as excess of blasts in bone marrow (5-19%) in the context of chronic eosinophilic leukemia. Reactive eosinophilia includes among all allergic, drug-induced, infectious, gastrointestinal, respiratory, endocrine and rheumatologic diseases, solid malignancy, lymphoma and lymphocytic variant HES (L-HES), as well as vasculitis [3]. Eosinophilic granulomatous with polyangiitis (EGPA) is the hallmark vasculitis presenting with eosinophilia. It is a small-vessel vasculitis mainly involving upper-airways and lungs (asthma-like presentation), heart, nervous system (mono/polyneuropathy), skin, musculoskeletal and gastrointestinal systems and may rarely lead to pauci-immune glomerulonephritis [4]. Primary results have shown a similar molecular basis of HES and EGPA which may actually represent the same disease [5]. Cardiac involvement from EGPA may resemble eosinophilic endo-myocarditis and is generally accompanied by high eosinophil counts and negative ANCA [6]. Therefore, and while clonal eosinophilia and most causes of reactive eosinophilia seemed to be ruled-out in our patient, some vasculitis/EGPA components were still present.
On the other hand, we cannot exclude that most of the aspects of our patient’s presentation may have been the expression of organ involvement from HES itself. Indeed, the non-specific rash that was detected has been described as a frequently encountered dermatologic involvement of HES mostly seen in the L-HES [7]. Cardiac involvement of HES is characterized by a 3-phase endo-myocarditis [8]. The first acute necrosis phase is clinically silent and detectable only by MRI findings of early stage myocardial eosinophilic inflammation but may manifest as subungual splinter hemorrhages representing small emboli from the endocardial surface [9]. Therefore, the splinter hemorrhages and corresponding capillaroscopy findings in our patient may be explained by the cardiac involvement of the disease. The disappearance of these findings subsequently may have been related to the disease course rather than to a vasculitis component.
The second stage of eosinophilic cardiac manifestation involves thrombus formation along areas of damaged myocardium, which were probably the source of emboli to the brain. This phase explains the MRI findings and elevated cardiac biomarkers and ECG changes observed in our patient. Finally, the third phase of cardiac involvement is characterized by restrictive cardiomyopathy due to endomyocardial fibrosis with generally irreversible MR which was observed in our patient [8]. Eosinophilia may as well lead to a hypercoagulable state due to the eosinophils’ storage and release of tissue factor initiating coagulation, and of cationic proteins activating platelets in thrombin generation and clot formation [10]. This may explain the thrombus formation observed in our patient and the positive test for lupus anticoagulant that normalized after the “thrombotic storm” that was experienced in the period of high disease activity. Neurologic involvement of HES is mainly related to the cardiac involvement and the thrombophilia since the main presentation is of cerebral embolization of cardiac origin [10]. This was observed in our patient that presented with recurrent cerebral emboli despite therapeutic anticoagulation. However, the pulmonary presentation of our patient was unusual. Indeed, a review of different lung involvements of HES among 49 patients related cases of patchy ground-glass opacities and consolidation, pulmonary nodules, asthma (including newly diagnosed) but no case of alveolar hemorrhage [11]. On the other hand, EGPA may present as alveolitis leading to alveolar hemorrhage, unrelated to eosinophilia end-organ damages [4]. Therefore, it is unclear if this was the first case of alveolar hemorrhage as a lung involvement of HES or a rare presentation of EGPA that included both hypereosinophilia and alveolar hemorrhage, without any other solid evidence of vasculitis.
Emergent treatment of HES is given with no respect to the cause of eosinophilia with high dose glucocorticoids in patients with extremely elevated eosinophil counts (above 100x109/L), signs/symptoms of leukostasis or life-threatening organ-involvement as was observed in our patient [7]. Eosinophil counts are supposed to drop within 48 hours of high dose glucocorticoids, unless the disease is steroid-unresponsive, which was the case in our patient. A review of steroid responsiveness of the different HES subtypes showed that all single organ, idiopathic, EGPA and lymphocytic variant HES eventually responded to glucocorticoid therapy in different doses, while the only HES subtype that was completely unresponsive was the myeloid variant HES [12].
Although no clonality was proved in our case, we still suspect there was some myeloproliferative components in this steroid-resistant HES. Some genetic changes may still be discovered, and our patient might present with one of these. Imatinib that was given to our patient only for a short period may be effective in some forms of M-HES, hydroxyurea decreases the eosinophil count by cytotoxicity so it is not specific to the HES subtypes, mepolizumab acts as an anti-IL5 and is effective in both clonal and reactive eosinophilia, cyclophosphamide is a chemo-immunotherapy that is particularly effective in the context of EGPA [7, 13-15]. Since our patient received all of these therapies in a short timing, we cannot be sure which of them were actually effective. However, since she presented with life-threatening signs of both vasculitis/reactive eosinophilia (alveolar hemorrhage) and clonal eosinophilia (steroid unresponsiveness), therapy (at least initially) with all of these agents seem to be the best approach until new clinical data are added to the case [16].
Conclusion
In conclusion, HES still presents difficulties in both diagnosis and management, while it may present with potentially life-threatening complications. Despite HES diagnostic uncertainties, treatment aggressiveness must fit its clinical and laboratory severity. Better understanding of its pathophysiology and further research on new cytogenetic changes leading to clonal eosinophilia are essential for the treatment approach in order to prevent severe and irreversible sequelae.
Conflicts of interest
None.
Article Info
Article Type
Case ReportPublication history
Received: Mon 25, Nov 2019Accepted: Fri 20, Dec 2019
Published: Thu 26, Dec 2019
Copyright
© 2023 Ilana Levy. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited. Hosting by Science Repository.DOI: 10.31487/j.JCMCR.2019.01.01
Figures & Tables
A) Splinter hemorrhages.
B) Unspecified rash.
C) Chest CT showing bilateral ground glass opacities suspicious for alveolar hemorrhage.
D) Brain MRI showing occipital infarcts with microhemorrhages.
E) Cardiac MRI showing multiple left ventricular thrombi.
F) Cardiac MRI showing diffuse subendocardial delayed enhancement in a distribution not fitting to a coronary territory.
Eos: Eosinophils; LAC: Lupus anticoagulant; MR: Mitral regurgitation; NBTE: Non-bacterial thrombotic endocarditis; PE: Plasma exchange; PHTN: Pulmonary hypertension; WBC: White blood cells.
References
- Simon HU, Rothenberg ME, Bochner BS, Peter F. Weller, Andrew J. Wardlaw et al. (2010) Refining the definition of hypereosinophilic syndrome. J Allergy Clin Immunol 126: 45-49. [Crossref]
- Kahn JE, Groh M, Lefèvre G (2017) (A Critical Appraisal of) Classification of Hypereosinophilic Disorders. Front Med (Lausanne) 4: 1-6. [Crossref]
- Gotlib J (2012) World Health Organization-defined eosinophilic disorders: 2012 update on diagnosis, risk stratification, and management. Am J Hematol 87: 903-914. [Crossref]
- Jagadeesh LY, Sangle SR, Verma H, D’Cruz D (2014) Alveolar haemorrhage in eosinophilic granulomatosis and polyangiitis (Churg-Strauss). Clin Rheumatol 33: 1177-1179.
- Baldini C, Galimberti S, Ciabatti E (2017) Extensive analysis of T cell receptor gamma (TCRG) gene rearrangements reveals a similar repertoire in eosinophilic granulomatosis with polyangitis (EGPA) and in hypereosinophilic syndrome (HES). Ann Rheum Dis.
- Bluett R, McDonnell D, O’Dowling C, Vaughan C (2017) Eosinophilic myocarditis as a first presentation of eosinophilic granulomatosis with polyangiitis (Churg-Strauss syndrome). BMJ Case Rep. [Crossref]
- Butt NM, Lambert J, Ali S, Beer PA, Cross NC, Duncombe A et al. (2017) Guideline for the investigation and management of eosinophilia. Br J Haematol 176: 553-572. [Crossref]
- Weller PF, Bubley GJ (1994) The Idiopathic Hypereosinophilic Syndrome. Blood 83: 2759-2779. [Crossref]
- Usui S, Dainichi T, Kitoh A, Miyachi Y, Kabashima K (2015) Janeway lesions and splinter hemorrhages in a patient with eosinophilic endomyocarditis. JAMA Dermatology 151: 907-908. [Crossref]
- Gotlib J, Cools J, III JMM, Schrier SL, Gilliland DG et al. (2004) The FIP1L1-PDGFRalpha fusion tyrosine kinase in hypereosinophilic syndrome and chronic eosinophilic leukemia: implications for diagnosis, classification, and management. Blood 103: 2879-2891. [Crossref]
- Dulohery MM, Patel RR, Schneider F, Ryu JH (2011) Lung involvement in hypereosinophilic syndromes. Respir Med 105: 114-121. [Crossref]
- Khoury P, Abiodun AO, Holland-Thomas N, Fay MP, Klion AD (2018) Hypereosinophilic Syndrome Subtype Predicts Responsiveness to Glucocorticoids. J Allergy Clin Immunol Pract 6: 190-195. [Crossref] .
- Weyand AC, Yanik GA, Bailey NG, Wu Y-M, Mody RJ et al. (2016) Imatinib Treatment in PDGFRA-Negative Childhood Hypereosinophilic Syndrome. Pediatr Blood Cancer 63: 164-167. [Crossref]
- Khoury P, Desmond R, Pabon A, Holland-Thomas N, Ware JM et al. (2016) Clinical features predict responsiveness to imatinib in platelet-derived growth factor receptor-alpha-negative hypereosinophilic syndrome. Allergy 71: 803-810. [Crossref]
- Klion AD (2015) How I treat hypereosinophilic syndromes. Blood 126: 1069-1077. [Crossref]
- Jin JJ, Butterfield JH, Weiler CR. Hematologic Malignancies Identified in Patients with Hypereosinophilia and Hypereosinophilic Syndromes. J Allergy Clin Immunol Pract 3: 920-925. [Crossref]