A Rare Case of Solitary Retroperitoneal Neurofibroma Mimicking Carcinoma of the Adrenal Gland
A B S T R A C T
Neurofibroma, a benign tumor of neural origin, indicates a possible differential diagnosis of adrenal masses in rare cases. It can be solitary or associated with neurofibromatosis type 1. Neurofibroma in the visceral compartment is an uncommon location and considered extremely rare. While it has a very low potential for malignant transformation, accurate diagnosis is nonetheless essential to allow for adequate treatment. In this article, we report a case of a patient with right-sided abdominal pain, fatigue and B symptoms (night sweats and weight loss) as a result of an adrenal tumor measuring 12 cm. The tumor was highly suspicious for malignancy, but the surgical resection revealed a benign neurofibroma. This case report describes a diagnostic and treatment pathway for nonspecific adrenal lesions.
Keywords
Neurofibroma, adrenal incidentaloma, retroperitoneal mass
Introduction
A neurofibroma located near the adrenal gland in the retroperitoneal space has been described as a very rare finding and only a few cases have been reported in the literature. Neurofibroma is a benign tumor of the peripheral nerves. It can be a solitary lesion or associated with the genetic disorder neurofibromatosis type-1 (NF-1). It develops commonly on the nerves in the skin, but in rare cases occurs in the abdominal cavity and can result in complications such as obstructive jaundice, gastrointestinal bleeding, or colonic intussusception [1, 2]. In this report, we describe a rare case of a retroperitoneal neurofibroma mimicking features of carcinoma of the adrenal gland. Although an extensive preoperative workup was performed, only the surgical resection revealed an accurate diagnosis.
Case Report
A 43-year-old woman was referred to our hospital for an assessment of a large mass identified in the right lumbar region on the computed tomography (CT) scan. The patient suffered from persistent right-sided lumbar pain for the past year, alongside fatigue, night sweats, intermittent palpitations and loss of appetite resulting in a 10 kg weight loss within the past 6 months. No illnesses, previous operations or long-term use of medication were reported.
On assessment, the patient appeared in good condition with normal vital signs. The patient exhibited right costovertebral angle tenderness on physical examination, but there was no palpable mass or clinical findings indicative of Cushing’s syndrome. The remainder of the examination was unremarkable. Routine laboratory tests of a complete blood count and C-reactive protein were normal. Measurements of plasma-free metanephrines, serum and urine cortisol, and androgens were within the normal ranges and excluded hormonal activity of the mass. Additional dexamethasone test showed appropriate suppression. Echinococcal serology was negative.
In addition to the written CT report, which was missing the imaging files, we ordered a magnetic resonance imaging (MRI) and a whole-body 18F-fluorodeoxyglucose–positron-emission tomography-CT (FDG-PET-CT) scan. The MRI confirmed a 12 cm mass in the right adrenal gland with heterogeneous enhancement and multiple necrotic areas (Figure 1). The mass displaced the right lobe of the liver and the right kidney. The inferior vena cava was partially compressed, but adjacent organs were intact with no signs of infiltration. The PET-CT scan characterized an adrenal lesion with an intense FDG uptake, but no FDG-avid lymph nodes or distant metastasis (Figure 2). Based on the morphology of the tumor, the radiologist reported a suspected carcinoma of the right adrenal gland.

Figure 1: MRI of the upper abdomen: Findings are compatible with adrenal carcinoma. No signs of infiltration of liver, right kidney or the inferior vena cava. a) axial T1-weighted image, b) coronal T2-weighted image.
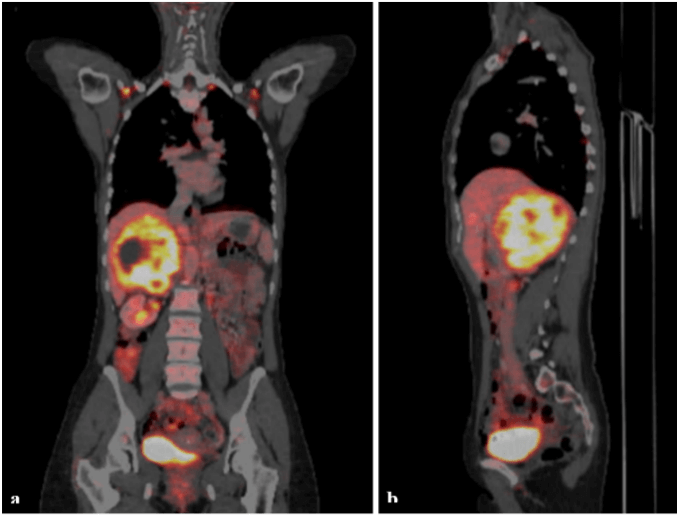
Figure 2: PET-CT scan: Tumor of the right adrenal gland with intense FDG-uptake. a) coronal plan, b) sagittal plane.
Although the physical examination, laboratory and imaging findings did not determine the definitive characteristics of the tumor, the patient’s medical history-specifically the B symptoms-pointed towards a malignancy. Based on the findings, we highly suspected adrenal cancer, and as recommended by the European guidelines, we abstained from a needle biopsy to avoid tumor cell spread and continued with an open surgical resection [3]. We performed a laparotomy through a right-subcostal incision with extension into the 10th intercostal space. Exploration of the abdomen disclosed a large tumor with a smooth surface and a repressing growth pattern. We gradually separated the tumor from the inferior vena cava by gentle, precise dissection, and performed an en-bloc resection of the tumor without capsular violation (Figure 3). Adjacent organs were apparently not infiltrated by the tumor and did not require surgical resection.
Histopathological examination revealed a large neurofibroma, with normal adrenal tissue adhered to the tumor (Figure 4). There was no infiltrative growth pattern nor any sign of malignancy. Neurofibroma generally suggests a correlation with NF-1, but the patient had no family history for NF-1 or other genetic syndromes, and the relevant medical history and physical examination revealed no signs of NF-1 or NF-2.
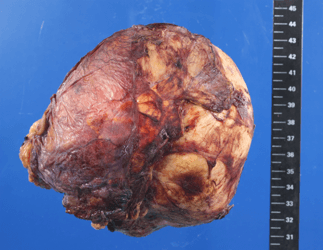
Figure 3: Surgical specimen: Well-circumscribed yellowish-brown tumor surrounded by a delicate capsule, measuring 12 cm by 12 cm by 8 cm.
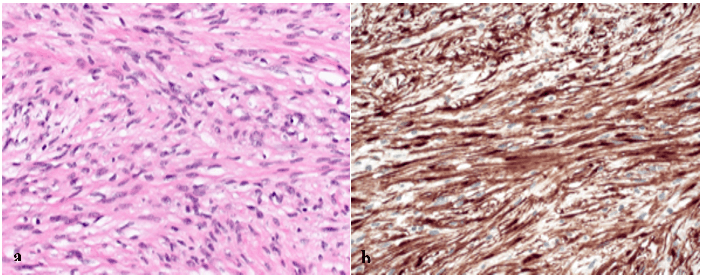
Figure 4: a) Histology (Hematoxylin and eosin, original magnification x200): Interlacing fascicles of elongated or plump spindle cells with cytologically bland wavy nuclei in collagenous matrix, b) Immunohistochemistry (S-100, original magnification x200): Strong positivity for S-100.
The patient developed a temporary adrenal insufficiency postoperatively, which was substituted. The postoperative course was uneventful, and the patient was discharged on the seventh postoperative day. Three weeks after the surgery, the patient had fully recovered, and the adrenal gland returned to normal function. A year later, the patient shows no clinical signs of recurrence or possible new neurofibromas.
Discussion
In this case, the origin and differentiation of the adrenal mass remained unclear even after thorough history taking, physical examination, laboratory and imaging diagnostics. Preoperative findings pointed strongly towards a malignancy; however, the definitive pathology workup revealed a benign neurofibroma. Adrenal tumors are commonly benign (69%) [4]. To determine the characteristics of the tumor, the guidelines recommend evaluating the size of the mass, its relation to adjacent structures, and its lipid content (expressed in Hounsfield units, HU) on a non-contrast CT scan [3]. Adrenal tumors that are <10 HU are very likely to be benign and require no further diagnostic imaging. If the findings remain unclear, then an MRI can be performed.
Masses that are >1 cm require further laboratory testing to assess the hormonal activity. These laboratory tests include cortisol, metanephrines, sex hormones, and aldosterone/renin ratio. Finally, a detailed patient and family history is recommended to exclude cancer and genetic disorders. European guidelines recommend against a needle biopsy for adrenal tumors to prevent the spread of tumor cells. A biopsy can be performed to exclude adrenal metastasis if the patient has a known cancer elsewhere. Surgical resection with en-bloc adrenalectomy is the standard approach for unilateral tumors. Open adrenalectomy is indicated for non-functional tumors with a >6 cm diameter to minimize the risk of capsular lesions, with the exception of myelolipoma [3, 5].
Neurofibromas are benign peripheral nerve sheath tumors and occur as solitary lesions or in association with NF-1. They consist of Schwann cells, fibroblasts and perineurial cells embedded in a collagenous matrix. The most common sites of solitary neurofibromas are the skin, peripheral nerves of subcutaneous or deeper body layers, and paraspinal nerve roots [6]. Retroperitoneal pararenal localization is very rare, especially in patients without NF-1.
In literature, we identified only a few individual case reports and a small case-series of 7 patients [7-9]. Leading symptoms included pain due to compression of adjacent structures, but B symptoms were not reported in other articles. Some authors performed a preoperative biopsy and isolated tumor resection, whereby others conducted a total nephrectomy without preoperative histopathological examination [7]. Definitive diagnosis was based on histopathological and immunohistochemical examinations [10]. Surgical resection requires an en-bloc resection without capsular violation and is often complicated by the tumor's dimension and vascularization. In conclusion, neurofibroma should be considered as a rare cause for abdominal pain and indicate a possible differential diagnosis of an adrenal mass. Currently, there are no standardized treatment guidelines for retroperitoneal neurofibroma. However, in the case of symptomatic adrenal tumors of uncertain malignancy, complete surgical resection should be performed.
Conflicts of Interest
None.
Funding
None.
Article Info
Article Type
Case ReportPublication history
Received: Mon 06, Apr 2020Accepted: Tue 21, Apr 2020
Published: Fri 24, Apr 2020
Copyright
© 2023 Loriana Lisarelli. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited. Hosting by Science Repository.DOI: 10.31487/j.JSCR.2020.02.05
Figures & Tables
References
- Bechade D, Boulanger T, Palazzo L, Algayres JP (2010) Primary neurofibroma of the common bile duct. Gastrointest Endosc 72: 895-897. [Crossref]
- Abramson LP, Orkin BA, Schwartz AM (1997) Isolated colonic neurofibroma manifested by massive lower gastrointestinal bleeding and intussusception. South Med J 90: 952-954. [Crossref]
- Fassnacht M, Arlt W, Bancos I, Dralle H, Newell-Price J et al. (2016) Management of adrenal incidentalomas: European Society of Endocrinology Clinical Practice Guideline in collaboration with the European Network for the Study of Adrenal Tumors. Eur J Endocrinol 175: G1-G34. [Crossref]
- Iniguez-Ariza NM, Kohlenberg JD, Delivanis DA, Hartman RP, Dean DS et al. (2017) Clinical, Biochemical, and Radiological Characteristics of a Single-Center Retrospective Cohort of 705 Large Adrenal Tumors. Mayo Clin Proc Innov Qual Outcomes 2: 30-39. [Crossref]
- Caek D (2019) S2k Guidelines: Resection strategies for adrenal tumours. AWMF.
- Bastounis E, Asimacopoulos PJ, Pikoulis E, Leppaniemi AK, Aggouras D et al. (1997) Benign retroperitoneal neural sheath tumors in patients without von Recklinghausen's disease. Scand J Urol Nephrol 31: 129-136. [Crossref]
- Corbellini C, Vingiani A, Maffini F, Chiappa A, Bertani E et al. (2012) Retroperitoneal pararenal isolated neurofibroma: report of a case and review of literature. Ecancermedicalscience 6: 253. [Crossref]
- Gupta P, Aggarwal R, Sarangi R (2015) Solitary neurofibroma of the adrenal gland not associated with type-1 neurofibromatosis. Urol Ann 7: 124-126. [Crossref]
- Falahatkar S, Mohammadzadeh A, Nikpour S, Khoshrang H, Askari K (2007) First reported case of adrenal neurofibroma in Iran. Urol J 4: 242-244. [Crossref]
- Dabkowski K, Marlicz W, Kaseja K, Sawicki M, Waloszczyk P et al. (2017) Solitary retroperitoneal neurofibroma: not as small as it seems. Pol Arch Intern Med 127: 701-702. [Crossref]