A Case Report on Melanotic Neuroectodermal Tumor of Infancy and Review of the Literature
A B S T R A C T
Melanotic neuroectodermal tumor of infancy (MNTI) is a rare, benign neoplasm of neural crest origin that most commonly occurs in the first year of life. The craniofacial area is affected 90% of the time with the maxilla being the affected site in the majority of cases. It has a recurrence rate of up to 27% and is associated with malignant transformation in up to 6.5% of patients. The current case describes the clinical presentation and management of a 3-month old male infant with MNTI of the maxilla. Following initial presentation, the tumor grew rapidly to compromise the aerodigestive tract. Surgical resection was performed and the adjacent bone was burred. There has been no clinical recurrence over a 4-year period of follow up.
Keywords
Melanotic neuroectodermal tumor of infancy, benign pigmented maxillary mass, neural crest tumor in the head and neck
Introduction
This rare, predominantly benign tumor was first described in 1918 by Krompecher and less than 500 cases have since been reported in the scientific literature [1, 2]. The cell of origin remained elusive for decades. Accordingly, the name was changed several times to reflect the cell that the tumor was thought to originate from at that time. However, after discovering that the tumor produced catecholamines, it was finally given the designation of melanotic neuroectodermal tumor of infancy as it was derived from neuroendocrine cells [1, 2]. The head and neck region is the primary site in over 90% of cases [3]. A rapidly growing, painless mass in the maxilla - as occurred with the patient in this case report is the most common presentation [4]. The tumor is more likely to occur in patients less than one year of age and malignant change can develop in up to 6.5% of these cases [4, 5]. Histology and immunohistochemistry are used to confirm the diagnosis [3, 6]. Complete surgical resection is the treatment of choice for MNTI [7]. Chemotherapy and radiotherapy however can have a role in patient management [8, 9]. Tumor recurrence occurs in up to 27% of patients and is less likely to occur if complete surgical resection was initially obtained [5, 10].
Case Report
This is the report of a 3-month-old male, who was referred to the San Fernando General Hospital (Trinidad, West Indies) for investigation of a painless mass in the upper jaw. The antenatal and birth history were uneventful. The infant was born at term via an uncomplicated vaginal delivery and was discharged together with his mother 24 hours after birth. The parents noticed a small maxillary swelling approximately ten days postnatally but did not think this was abnormal. The mass gradually increased in size over the next three weeks which prompted them to seek medical attention. There was no history of trauma or insect bites nor was there a recent travel history. The patient was first taken to a doctor at a private facility where the parents were advised that the child needed imaging and a biopsy. However, they could not afford this and was referred to the public hospital. The parents then decided to observe the child for possible regression of the mass for a few weeks but when it continued growing, they finally brought the child to the hospital.
The patient was seen by the Otolaryngology Department at 3-months of age. There was no associated difficulty in breathing or feeding. A general examination revealed right facial asymmetry. The right nostril was distorted and displaced upwards. There was no nasal discharge nor was an intranasal mass detected. An oral examination revealed a 5 x 4 cm smooth, firm, non-friable mass in the anterior aspect of the maxilla (Figure 1). It was light pink in colour and was more prominent on the right side. The deciduous teeth were unerupted. The ear and neck exam were normal.
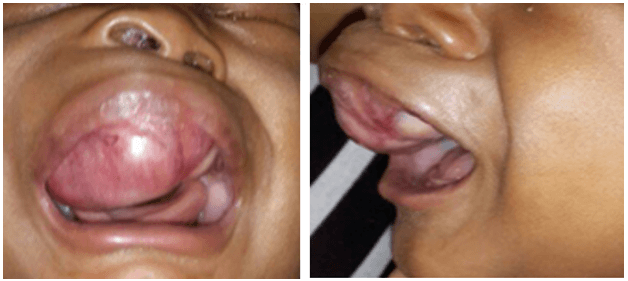
Figure 1: A well circumscribed, non-pigmented maxillary mass – AP and lateral views.
A fine needle aspiration cytology specimen of the mass was obtained. However, the samples were contaminated with blood and had to be discarded. An open biopsy was then attempted but this had to be abandoned due to significant bleeding. A CT scan of the facial bones with contrast revealed a well-demarcated heterogenous lesion in the right maxilla with expansion and thinning of the adjacent bone (see arrows in Figure 2). The lesion also exhibited mild enhancement with intravenous contrast. The possibility of a haemangioma was entertained and it was decided that the lesion would be observed in order to assess its clinical behaviour before attempting further investigations or starting propranolol. The parents were given a 2-month clinic appointment with advice to return to hospital earlier if there were any significant changes.
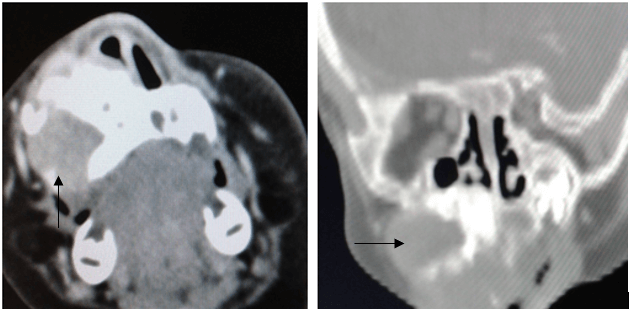
Figure 2: A CT scan of the facial bones with contrast. Mass indicated by black arrows on both A) axial and B) coronal views.
At the first follow-up appointment, the parents reported no difficulty in breathing and the mother stated that she was still able to feed the child with a bottle. The mass bled intermittently but the episodes were mild and self-limiting. Therefore, they had decided to wait until the scheduled clinic appointment to see a doctor despite the rapid growth of the lesion. On examination, the mass was noted to have undergone significant enlargement and now demonstrated patches of dark pigmentation as seen in (Figure 3). Therefore, the diagnosis was revised to that of mucosal melanoma and the decision made to perform an excision biopsy in order to confirm the diagnosis as well as to circumvent aerodigestive complications. The patient could not be intubated despite several attempts (including fibreoptic intubation) and had to be sedated with a bag and mask while a tracheostomy was performed. Once the airway was secured, a Weber Ferguson incision was made to expose the involved facial bones. This allowed for an assessment of the extent of tumor involvement to be made. The mass extended superiorly to within 1cm of the right infraorbital rim. The tumor was excised without attention to margins; preserving the infraorbital rim and orbital floor. Unerupted teeth within the specimen were also removed. The surrounding bone was then removed with a burr until the erosive bony de-cortification as shown by the black arrow in (Figure 4) and pigmented areas as indicated by the blue arrow in (Figure 4) were no longer observed.
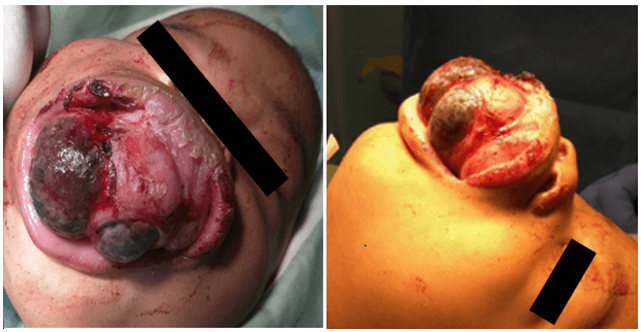
Figure 3: The appearance of the maxillary mass at five months of age – A) AP and B) lateral views.

Figure 4: Surgical removal of right maxillary tumor.
Histological examination of the specimen under low power magnification revealed clusters of basophilic neuroblast-like cells along with melanocyte-like cells arranged in alveolar or pseudoglandular structures (Figure 5A). A more high-powered view demonstrated vesicular nuclei and variable amounts of melanin pigment within melanocyte-like cells (Figure 5B). The final histological diagnosis was a melanotic neuroectodermal tumor of infancy of the right maxilla.

Figure 5: A) Low power magnification (x4), B) High power magnification (x40).
The patient has since been followed up in the outpatient’s clinic (Figures 6-8) every 3 months for the first 2 years and every 4 months for the next two years. There has been no clinical evidence of local recurrence and the neck remains clear of disease. A right facial asymmetry was noted at the age of 3 (Figure 7). A repeat CT scan of the facial bones was performed which reported a lesion in the right maxilla that was non-specific for recurrence and may have represented reactive surgical changes. The parents refused further surgical intervention but agreed to continue with follow up. At four years of age, the patient remains clinically free of tumor (Figure 8).
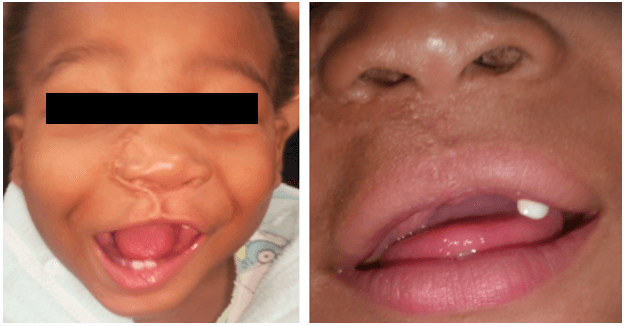
Figure 6: Patient at 2 years of age.

Figure 7: Patient at 3 years of age.
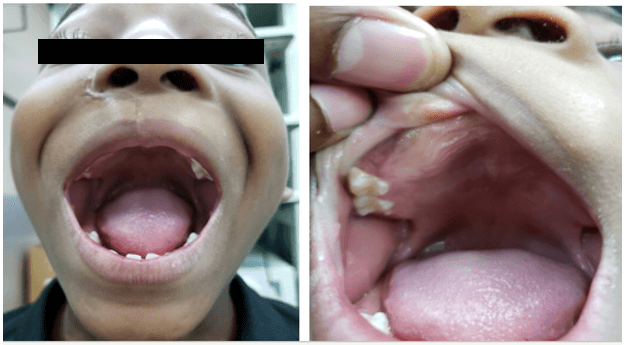
Figure 8: Patient at 4 years of age.
Discussion
Melanotic neuroectodermal tumor of infancy (MNTI) usually affects patients that are less than one year of age but prenatal and congenital cases have been reported [11-13]. The condition has also occurred in older children and adults in a minority of cases [14, 15]. Most publications have stated no gender predilection whilst a few have noted a slight male predominance [6, 16]. It primarily develops in the head and neck region with the maxilla being the most common anatomical site affected [3, 4]. In most cases, the tumor develops within the soft tissue overlying the maxilla which then rapidly grows into and destroys the underlying bone [7, 14]. A variety of local and regional sequelae may ensue including decreased oral functionality and aerodigestive tract compromise [7]. If left untreated, long term effects such as abnormal craniofacial, dental and orthodontic development as well as speech problems can also occur. These can all contribute to psychological and social issues.
Other reported sites of origin include the central nervous system, mediastinum, uterus, epididymis, ovary, testes, upper arm, thigh, post auricular subcutaneous tissue and skin [3, 17-21]. Malignant change can occur and has been reported to range from 1.9% – 6.5% of cases. There are no factors to predict this change in biologic behaviour [4, 6, 15, 22, 23]. This malignant variant spreads by direct invasion into adjacent tissue but can also metastasize to regional and distant sites such as lymph nodes, liver and bone [4]. Histologically, malignant MNTI demonstrates increased mitosis > 2 per 10 high power field, hypercellularity and focal necrosis [24-27]. The predominant cell type in metastatic deposits are the neuroblast-like cells [23, 24]. The tumor usually presents as a rapidly growing, non-ulcerating, painless, solitary mass [7, 20]. However, in 9% of cases, they can be multifocal [8, 17]. Pigmentation may not always be obvious in the tissues even though melanin is being produced as was the case initially for the patient in this case report [11, 22, 28].
Hematological tests for catecholamines and urinary testing for vanillymandelic acid can sometimes be elevated as a result of the neural crest origin of the tumor [25]. Levels should be obtained in order to assist with treatment planning but it has no influence on the biologic activity of the tumor. Therefore, these tests cannot be used to distinguish benign from malignant variants [5, 29]. Plain radiographs and ultrasound scans do not provide enough detail regarding the tumor and the involved bone. Therefore, cross sectional imaging in the form of computed tomography (CT) or magnetic resonance imaging (MRI) scans are preferred. On CT scanning, MNTI appears as a heterogenous, hyperdense lesion with bony expansion or destruction as was seen in the CT scan of our patient [30]. The MRI scan gives the best details of the tumor itself. An isointense or hypointense lesion is imaged on T1 and T2 sequences which enhances with the administration of contrast [16, 30]. Alternatively, T1 sequences can appear hyperintense if a large amount of melanin is present within the tumor [31]. An additional advantage that MRI has over CT is the lack of radiation exposure which allows the test to be repeatedly performed [16, 31].
Diagnosis of MNTI is confirmed by histology and immunohistochemistry. The classic histological appearance is of a dual population of neuroblast-like cells and epithelial cells within a fibrocollagenous stroma [3, 7]. This tumor does not have rosette formation. The neuroblastic cells appear as small round blue cells and the larger epithelial cells produce melanin [7, 21]. Immunohistochemical analysis is positive for cytokeratin, glial fibrillary acidic protein, vimentin, epithelial membrane antigen, neuron-specific enolase, human melanoma black 45, CD 56, c-myc, dopamine β-hydroxylase and synaptophysin [24, 27, 32]. MNTI is usually negative for CD99 or Ki-77 but in cases where it is positive - this may indicate more aggressive behaviour or a malignant variant [23, 27]. The tumor stains negative for chromogranin A, S100 and neurofilaments [33]. In some patients, alpha fetoprotein levels can also be elevated [24]. Molecular testing is relatively new and can help identify mutations within the lesion. The BRAFV600E, CDKN2A and RPLP1- C19MC fusion mutations have been noted in a few cases of MNTI [33, 34]. Translocations in t(11;22)(q24;q12) and t(11;22)(p13;q12) as well as MYCN gene amplification and the deletion of 1p have also been documented [35]. Molecular testing is not confirmatory. However, it can provide information regarding the sensitivity of individual tumors to chemotherapy [4].
Optimal management of MNTI involves early diagnosis combined with complete tumor resection and close follow up. Surgery is the mainstay of treatment and can be used alone or in combination with chemotherapy or radiotherapy [14]. Controversy exists regarding how much bone needs to be removed in order to achieve adequate resection margins given the infiltrative growth pattern of the tumor [3]. Resection with a 2-5mm margin is often recommended once there is no functional or developmental compromise [16, 24, 29]. In the maxilla or mandible, the involved or adjacent teeth are also removed [14]. In anatomical areas such as the orbital floor, skull and sometimes the mandible; it may not be possible to achieve these margins and a more conservative resection has to be performed [16]. For these cases, enucleation and curettage of the adjacent tissue can be considered. Close follow up is mandatory for these patients and further resection or chemoradiation should be considered if there is recurrence. Reconstruction is performed once the tumor is confirmed to be completely excised.
Rickart et al. in 2019 outlined their management protocol for MNTI [16]. Accordingly, resection with a 5mm margin is advocated once the tumor is well localized. Even though the lesion was localized in our patient, the working diagnosis was that of mucosal melanoma with bony involvement hence an excision biopsy was carried out. Additionally, the involved teeth were taken in the resection and the adjacent bone was removed until clinically normal bone architecture was observed. We had attained gross excision of the mass and burred the surrounding bone which should have produced similar results to those attained by curettage. The changes seen on CT scan when the patient was 3-years old could have been due to recurrent disease or reactive changes due to surgery. Clinically, the patient had no visible masses, discolouration or regional lymphadenopathy to indicate a recurrence. In addition, the parents did not want any further surgery. Therefore, observation continued without a repeat biopsy.
Other treatment options that have been used include chemoradiation, surgery with adjuvant chemoradiation, neoadjuvant chemotherapy followed by surgery and adjuvant chemotherapy [5, 8, 13, 16, 36, 37]. The rational for using neoadjuvant chemotherapy is to reduce the size of the tumor so that a more complete resection may be obtained. Neoadjuvant treatment is offered to patients with vital structure involvement or who may be ‘inoperable’ [37, 38]. Chemotherapy has been proposed to induce the maturation of the neuroblast-like cells thus reducing this cell population and increasing the amount of epithelial cells [3]. In cases of malignant MNTI, the prognosis is very poor. A combination of conservative tumor resection and chemotherapy is recommended as radical surgery has not been proven to change the outcome [7, 16].
Recurrence for non-malignant MNTI ranges from 10-27% [10, 15, 16]. Clinical recurrence is associated with tumor development at an age of less than 2 months. The tumor in our patient was noted within days after birth even though the official diagnosis was made after 2 months. This placed the patient at the highest risk of recurrence. An intermediate risk is present between 2.5 and 4 months of age [6, 39]. Most recurrences occur within 6 months of treatment [40]. However, no such recurrence has been noted in our patient after almost 4 years of follow up. Late recurrences are uncommon [4]. Also, not all patients with positive margins develop recurrence and this phenomenon was attributed to stimulation of the host immune response by surgical extirpation of the tumor. Close surveillance after surgery remains the best indicator of prognosis [24]. The majority of patients with MNTI can be cured with complete surgical resection [41]. However, the malignant variant continues to have a poor outcome.
Conclusion
Melanotic neuroectodermal tumors of infancy are rare, benign neoplasms of neural crest origin. Most occur in children under the age of one year. However, it has also been documented to occur in prenatal, congenital, older children and adult patients. The majority of cases occur in the head and neck region with the maxilla being the most commonly affected site. Malignant transformation can occur in up to 6.5% of patients. It presents as a rapidly growing, painless mass with variable degrees of pigmentation. Histological and immunohistochemical assessment confirms the diagnosis. Complete surgical resection to 5 mm margins is the treatment of choice. Chemotherapy and radiotherapy have a limited role in patient management. Tumor recurrence can occur in up to 27% of patients and is more likely to occur in patients diagnosed within the first two months of life.
Article Info
Article Type
Case Report and Review of the LiteraturePublication history
Received: Thu 25, Jun 2020Accepted: Mon 06, Jul 2020
Published: Fri 10, Jul 2020
Copyright
© 2023 Gailann Jugmohansingh. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited. Hosting by Science Repository.DOI: 10.31487/j.AJSCR.2020.03.06
Figures & Tables
References
- Borello ED, Gorlin RJ (1966) Melanotic neuroectodermal tumor of infancy--a neoplasm of neural crest origin. Report of a case associated with high urinary excretion of vanilmandelic acid. Cancer 19: 196-206. [Crossref]
- A P Tan, Thomas S Jacques, Kshitij Mankad, Gregory James, Owase Jeelani et al. (2018) Melanotic neuroectodermal tumour of infancy: A case report and differential diagnosis. Neuroradiol J 31: 434-439. [Crossref]
- Brian S Soles, Allecia Wilson, David R Lucas, Amer Heider (2018) Melanotic Neuroectodermal Tumor of Infancy. Arch Pathol Lab Med 142: 1358-1363. [Crossref]
- Birgit Kruse Lösler, Christoph Gaertner, Horst Bürger, László Seper, Ulrich Joos et al. (2006) Melanotic neuroectodermal tumor of infancy: systematic review of the literature and presentation of a case. Oral Surg Oral Med Oral Pathol Oral Radiol Endod 102: 204-216. [Crossref]
- Amit Chaudhary, Ashish Wakhlu, Neetu Mittal, Sanjeev Misra, Divya Mehrotra et al. (2009) Melanotic neuroectodermal tumor of infancy: 2 decades of clinical experience with 18 patients. J Oral Maxillofac Surg 67: 47-51. [Crossref]
- Audrey Moreau, Louise Galmiche, Veronique Minard Colin, Martin Rachwalski, Kahina Belhous et al. (2018) Melanotic neuroectodermal tumor of infancy (MNTI) of the head and neck: a French multicenter study. J Craniomaxillofac Surg 46: 201-206. [Crossref]
- Pooja Agarwal, Susmita Saxena, Sanjeev Kumar, Richi Gupta (2010) Melanotic neuroectodermal tumor of infancy: Presentation of a case affecting the maxilla. J Oral Maxillofac Pathol 14: 29-32. [Crossref]
- B Halpert, R Patzer (1947) Maxillary tumor of retinal anlage. Surgery 22: 837-841. [Crossref]
- Willi Woessmann, Markus Neugebauer, Regina Gossen, Renate Blütters Sawatzki, Alfred Reiter (2003) Successful chemotherapy for melanotic neuroectodermal tumor of infancy in a baby. Med Pediatr Oncol 40: 198-199. [Crossref]
- Saleh Rachidi, Amit J Sood, Krishna G Patel, Shaun A Nguyen, Heidi Hamilton et al. (2015) Melanotic neuroectodermal tumor of infancy: a systematic review. J Oral Maxillofac Surg 73: 1946-1956. [Crossref]
- Márcia Gaiger de Oliveira, Lester D R Thompson, Anna Ceclia Moraes Chaves, Pantelis Varvaki Rados, Isabel da Silva Lauxen et al. (2004) Management of melanotic neuroectodermal tumor of infancy. Ann Diagn Pathol 8: 207-212. [Crossref]
- M Koob, C Fayard, D Pariente, C Adamsbaum, S Franchi Abella (2015) Prenatal diagnosis of orbital melanotic neuroectodermal tumor in infancy. Ultrasound Obstet Gynecol 46: 249-250. [Crossref]
- Audrey Moreau, Louise Galmiche, Kahina Belhous, Gerald Franchi, Vincent Couloigner et al. (2018) Prenatal Diagnosis of a Melanotic Neuroectodermal Tumor of Infancy (MNTI): A Case Report With a Favorable Outcome After Chemotherapy Failure and Incomplete Resection. J Pediatr Hematol Oncol 40: 320-324. [Crossref]
- Tram Kiem H, Nguyen Thi KH, Tran Xuan P, Nguyen Hong L, Nguyen Huu S et al. (2019) Melanocytic neuroectodermal tumour of infancy. J Pediatr Surg Case Rep 46: 101221.
- L S Cutler, A P Chaudhry, R Topazian (1981) Melanotic Neuroectodermal tumor of infancy: An ultrastructural study. Literature review and re-evaluation. Cancer 48: 257-270. [Crossref]
- A J Rickart, V Drummond Hay, A Suchak, Z Sadiq, N J Sebire et al. (2019) Melanotic neuroectodermal tumour of infancy: Refining the surgical approach. Int J Oral Maxillofac Surg 48: 1307-1312. [Crossref]
- Pramod Jain, Rajeev Kumar Garg, Anjali Kapoor (2010) Melanotic neuroectodermal tumor of infancy in oral cavity at unusual age. Fetal Pediatr Pathol 29: 344-352. [Crossref]
- S Evans, A Woolley (2018) Postauricular Melanocytic Neuroectodermal Tumor of Infancy: A Rare Site of a Rare Tumor-MNTI as a Postauricular Mass with Literature Review. Case Rep Otolaryngol 2018: 9829856. [Crossref]
- Monica Stephany Kirigin, Tihana Džombeta, Sven Seiwerth, Marko Mesić, Jasminka Stepan Giljević et al. (2017) Melanotic Neuroectodermal Tumor of Infancy of the Upper Arm. Med Princ Pract 26: 582-585. [Crossref]
- Shanon R Lacy, Matthew Kuhar (2010) Melanotic neuroectodermal tumor of infancy presenting in the subcutaneous soft tissue of the thigh. Am J Dermatopathol 32: 282-286. [Crossref]
- Balaji Babu Bangi, M L Avinash Tejasvi (2012) Melanotic neuroectodermal tumor of infancy: a rare case report with differential diagnosis and review of the literature. Contemp Clin Dent 3: 108-112. [Crossref]
- Bansal A, Bansal R, Kumar K, Shetty D, Kapoor C (2010) Melanotic neuroectodermal tumor of infancy (MNTI): A case report with historical insights and review in relation to its origin. Int J Pathol 11.
- Kenjiro Higashi, Takenori Ogawa, Masaei Onuma, Hajime Usubuchi, Yoshimichi Imai et al. (2016) Clinicopathological features of melanotic neuroectodermal tumor of infancy: report of two cases. Auris Nasus Larynx 43: 451-454. [Crossref]
- Scott Hamilton, Duncan Macrae, Sumit Agrawal, Damir Matic (2008) Melanotic neuroectodermal tumour of infancy. Can J Plast Surg 16: 41-44. [Crossref]
- Asaranti Kar, Priyadarshini Biswal, Susmita Behera, Ipsita Dhal, Surabhi (2015) Cytologic Interpretation of Melanotic Neuroectodermal Tumour of Infancy Involving Cranial Bones: Clue to Diagnosis. J Clin Diagn Res 9: ED12-ED14. [Crossref]
- In Hyuk Yoo, Sook Kyung Yum, Se Jeong Oh, Kyoung Mee Kim, Dae Chul Jeong (2014) Melanotic Neuroectodermal Tumor of Infancy Disseminated by a Ventriculoperitoneal Shunt and Diagnosed from the Inguinal Sac. J Pediatr Hematol Oncol 36: e61-e64. [Crossref]
- A W Barrett, M Morgan, A D Ramsay, P M Farthing, L Newman et al. (2002) A clinicopathologic and immunohistochemical analysis of melanotic neuroectodermal tumor of infancy. Oral Surg Oral Med Oral Pathol Oral Radiol Endod 93: 688-698. [Crossref]
- R Ijiri, K Onuma, M Ikeda, K Kato, Y Toyoda et al. (2001) Pigmented intraosseous odontogenic carcinoma of the maxilla: A pediatric case report and differential diagnosis. Hum Pathol 32: 880-884. [Crossref]
- Y Hoshina, Y Hamamoto, I Suzuki, T Nakajima, H Ida Yonemochi et al. (2000) Melanotic neuroectodermal tumor of infancy in the mandible: Report of a case. Oral Surg Oral Med Oral Pathol Oral Radiol Endod 89: 594-599. [Crossref]
- C Suzuki, M Maeda, N Matsushima, M Takamura, T Matsubara et al. (2007) Melanotic neuroectodermal tumor of infancy in the skull: CT and MRI features. J Neuroradiol 34: 212-213. [Crossref]
- Saira Haque, Mary Beth McCarville, Neil Sebire, Kieran McHugh (2012) Melanotic neuroectodermal tumour of infancy: CT and MR findings. Pediatr Radiol 42: 699-705. [Crossref]
- Luciana Strieder, Román Carlos, Jorge Esquiche León, Alfredo Ribeiro Silva, Victor Costa et al. (2016) Protumorigenic M2-like phenotype cell infiltration in the melanotic neuroectodermal tumor of infancy. Oral Surg Oral Med Oral Pathol Oral Radiol 121: 173-179. [Crossref]
- Carolina C Gomes, Marina G Diniz, Grazielle Helena F de Menezes, Wagner H Castro, Ricardo S Gomez (2015) BRAFV600E Mutation in Melanotic Neuroectodermal Tumor of Infancy: Toward Personalized Medicine? Pediatrics 136: e267-e269. [Crossref]
- David J Barnes, Edward Hookway, Nick Athanasou, Takeshi Kashima, Udo Oppermann et al. (2016) A germline mutation of CDKN2A and a novel RPLP1-C19MC fusion detected in a rare melanotic neuroectodermal tumor of infancy: a case report. BMC Cancer 16: 629. [Crossref]
- M Khoddami, J Squire, M Zielenska, P Thorner (1998) Melanotic neuroectodermal tumour of infancy: a molecular genetic study. Pediatr Dev Pathol 1: 295-299. [Crossref]
- Johannus Neven, Christine Hulsbergen van der Kaa, Jacqueline Groot Loonen, Peter C M de Wilde, Matthais A W Merkx (2008) Recurrent melanotic neuroectodermal tumour of infancy: a proposal for treatment protocol with surgery and adjuvant chemotherapy. Oral Surg Oral Med Oral Pathol Oral Radiol Endod 106: 493-496. [Crossref]
- Christopher Maroun, Ibrahim Khalifeh, Elie Alam, Pierre Abi Akl, Raya Saab et al. (2016) Mandibular melanotic neuroectodermal tumor of infancy: a role for neoadjuvant chemotherapy. Eur Arch Otorhinolaryngol 273: 4629-4635. [Crossref]
- C Murphy, J Pears, G J Kearns (2016) Melanotic neuroectodermal tumour of infancy: surgical and chemotherapeutic management. Ir J Med Sci 185: 753-756. [Crossref]
- Abhishek Kumar, V Deepthi, Ruchi Aggarwal, Hema Malini Aiyer (2018) Melanotic neuroectodermal tumor of infancy: a rare case report. J Oral Maxillofac Pathol 22: S44-S47. [Crossref]
- S B Kapadia, D M Frisman, C L Hitchcock, G L Ellis, E J Popek (1993) Melanotic neuroectodermal tumor of infancy. Clinicopathological, immunohistochemical, and flow cytometric study. Am J Surg Pathol 17: 566-573. [Crossref]
- H H Liu, T W Chen, H S Chang (2004) Melanotic neuroectodermal tumor of infancy in the maxilla: a case report. Int J Paediatr Dent 14: 371-375. [Crossref]